1. 领域背景与文献引入
文献英文标题:PTK2 is a potential biomarker and therapeutic target for EGFR- or TLRs-induced lung cancer progression via the regulation of the cross-talk between EGFR- and TLRs-mediated signals;发表期刊:Biomarker Research;影响因子:未公开;研究领域:非小细胞肺癌(NSCLC)生物标志物与靶向治疗。
非小细胞肺癌(NSCLC)是肺癌的主要病理类型,占比约85%,其进展与表皮生长因子受体(EGFR)突变激活、toll样受体(TLRs)介导的炎症信号异常密切相关。现有研究证实,EGFR酪氨酸激酶抑制剂(TKIs)是EGFR突变NSCLC的一线治疗,但耐药性问题突出;TLRs激活通过NF-κB通路促进肿瘤微环境炎症,加速转移。蛋白酪氨酸激酶2(PTK2)作为黏着斑激酶家族成员,在多种肿瘤中高表达并促进细胞迁移,但PTK2与EGFR、TLRs信号通路的功能协同及临床关联尚未明确——这是领域内未解决的核心问题,直接限制了NSCLC精准治疗靶点的开发。针对这一空白,本研究通过整合临床样本分析、细胞功能实验、动物模型验证及靶向抑制剂研究,系统探究PTK2在EGFR或TLRs诱导NSCLC进展中的作用机制,旨在明确其作为生物标志物及治疗靶点的潜力。
2. 文献综述解析
作者对现有研究的评述逻辑以“单基因关联-未解决协同问题”为核心维度:一方面,现有研究已证实PTK2、EGFR或TLRs的单独表达异常与NSCLC的发生、转移及不良预后相关(如EGFR突变导致增殖信号持续激活,TLRs激活促进肿瘤炎症);另一方面,三者的功能协同及临床关联尚未被阐释——多数研究聚焦于单一通路的调控机制,缺乏对“PTK2-EGFR-TLRs”信号网络的整合分析,且尚未通过功能实验验证三者的相互作用。
现有研究的关键结论包括:PTK2在肿瘤中高表达并促进细胞迁移,EGFR-TKIs治疗EGFR突变NSCLC但耐药性常见,TLRs激活通过NF-κB促进肿瘤进展;技术方法上,现有研究多采用临床样本的基因表达谱分析,优势在于直接关联基因表达与临床结局,但局限性是缺乏细胞或动物水平的功能验证,无法明确因果关系。
本研究的创新价值在于首次将PTK2与EGFR、TLRs信号通路关联,通过“临床样本-细胞模型-动物实验-治疗验证”的完整链条,揭示PTK2通过调控EGFR与TLRs信号的交叉对话促进NSCLC进展的机制,弥补了现有研究“单一基因聚焦”的不足,为PTK2作为EGFR或TLRs诱导NSCLC的生物标志物及治疗靶点提供了直接的功能与临床证据。
3. 研究思路总结与详细解析
本研究以“临床关联-机制验证-治疗潜力”为核心逻辑,形成“临床样本分析→细胞功能验证→动物模型验证→治疗靶点确认”的闭环路线。研究目标是明确PTK2与EGFR、TLRs在NSCLC中的功能及临床关联,验证PTK2作为治疗靶点的潜力;核心科学问题是PTK2如何通过调控EGFR与TLRs信号的交叉对话促进NSCLC进展;技术路线涵盖“临床样本基因分析→CRISPR-Cas9细胞模型→体外功能实验→NSG小鼠异种移植→抑制剂治疗验证”。
3.1 临床样本基因表达与生存关联分析
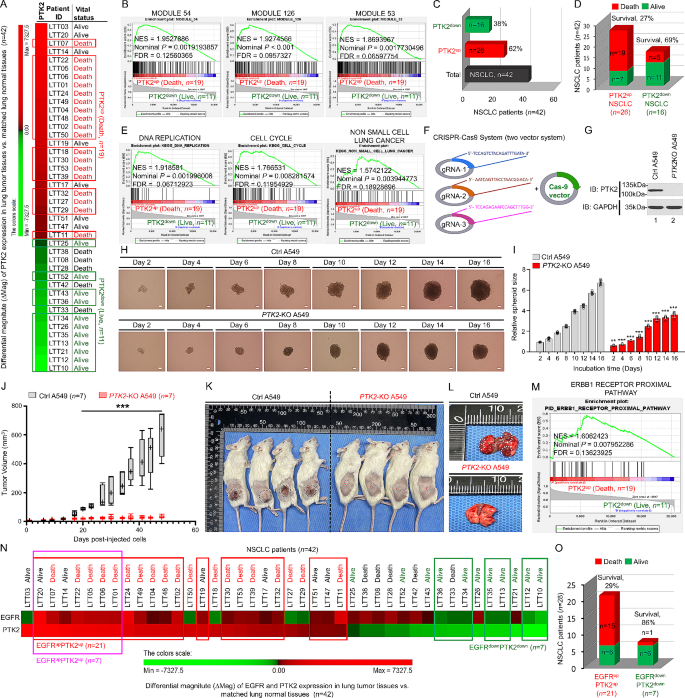
实验目的是明确PTK2、EGFR、TLRs在NSCLC患者中的基因表达差异及与生存预后的关联。方法细节:收集42例NSCLC患者的肿瘤组织及配对正常组织,通过微阵列技术检测基因表达水平,采用差分 magnitude(ΔMag)分析将患者分为PTK2高表达(PTK2up)与低表达(PTK2down)组,结合患者临床结局(生存状态、肿瘤进展)进行基因集富集分析(GSEA)及生存曲线绘制。结果解读:PTK2up患者(n=19,死亡)的肿瘤组织中,“癌症模块”“细胞周期”“NSCLC进展”相关基因集显著富集(GSEA显示归一化富集得分(NES)>1.5,FDR<0.05);PTK2up组患者的总生存期显著短于PTK2down组(PTK2up组n=26,中位生存期约18个月;PTK2down组n=16,中位生存期约36个月,P<0.01);EGFR与PTK2共高表达(EGFRupPTK2up)患者的生存百分比显著低于共低表达患者(29% vs 86%,n=21 vs n=7,P<0.001)。产品关联:文献未提及具体实验产品,领域常规使用基因表达微阵列芯片(如Affymetrix Human Genome U133 Plus 2.0 Array)及GraphPad Prism生存分析软件。
3.2 PTK2敲除细胞模型构建与验证
实验目的是构建PTK2基因敲除的肺癌细胞系,为后续功能实验提供工具。方法细节:采用CRISPR-Cas9基因编辑技术,设计3条针对PTK2基因的gRNA,通过双载体系统(Cas9表达载体与gRNA载体)转染A549(肺腺癌细胞)与H1299(肺鳞癌细胞)细胞,经嘌呤霉素筛选获得稳定敲除细胞系(PTK2-KO);通过免疫印迹(WB)实验验证PTK2蛋白的敲除效率。结果解读:WB结果显示,PTK2-KO细胞系中PTK2蛋白表达几乎完全消失(与对照细胞相比,条带灰度值降低>90%),成功构建功能缺失的细胞模型。产品关联:实验所用关键技术为CRISPR-Cas9基因编辑,领域常规使用gRNA合成试剂盒(如IDT gRNA Synthesis Kit)、Cas9表达载体(如pSpCas9-2A-Puro)。
3.3 体外肿瘤进展功能验证
实验目的是验证PTK2敲除对肺癌细胞增殖、迁移、3D肿瘤球形成的影响。方法细节:①细胞增殖实验:采用CCK-8法检测PTK2-KO与对照细胞的增殖能力(培养0、24、48、72小时);②细胞迁移实验:通过Transwell小室检测细胞跨膜迁移能力(培养24小时后计数迁移细胞数);③3D肿瘤球形成实验:将细胞接种于琼脂糖水凝胶包被的96孔板,培养48小时后观察肿瘤球形成,通过ImageJ软件定量肿瘤球大小(n=7)。结果解读:PTK2-KO细胞的增殖速率显著低于对照细胞(如A549细胞72小时增殖率降低约40%,P<0.01);迁移细胞数减少约50%(P<0.001);3D肿瘤球的大小显著缩小(PTK2-KO A549细胞肿瘤球体积较对照减少约60%,n=7,P<0.001),表明PTK2缺失显著抑制肺癌细胞的体外恶性表型。产品关联:文献未提及具体实验产品,领域常规使用CCK-8试剂盒(如Dojindo CCK-8)、Transwell小室(如Corning Transwell)、ImageJ图像分析软件。
3.4 体内异种移植模型验证
实验目的是验证PTK2敲除对体内肿瘤生长及转移的抑制作用。方法细节:将PTK2-KO或对照A549细胞(5×10⁶个/只)皮下注射到NSG小鼠背部(每组n=7),每3天测量肿瘤体积(公式:(长×宽)²×0.5),48天后处死小鼠,观察肺转移情况。结果解读:PTK2-KO组小鼠的肿瘤生长速率显著慢于对照组(48天肿瘤体积约为对照组的30%,P<0.001);对照组小鼠肺组织可见明显转移灶(白色结节),而PTK2-KO组几乎无转移灶。产品关联:实验所用动物模型为NSG(NOD/SCID/IL-2Rγnull)小鼠(购自Jackson Laboratory);肿瘤体积测量使用游标卡尺(如Mitutoyo卡尺)。
3.5 PTK2与EGFR/TLRs信号相互作用机制解析
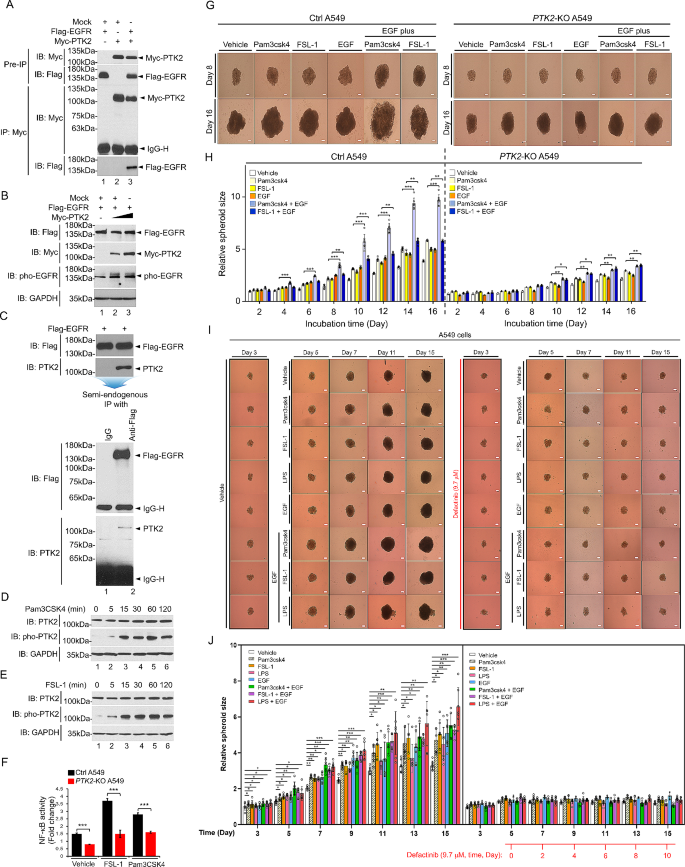
实验目的是探究PTK2调控EGFR与TLRs信号交叉对话的分子机制。方法细节:①免疫沉淀(IP)实验:在HEK-293T细胞中过表达Flag-EGFR与Myc-PTK2,或在A549细胞中过表达Flag-EGFR,通过IP检测PTK2与EGFR的相互作用;②免疫印迹(WB)实验:检测PTK2-KO或对照细胞在TLR agonists(Pam3CSK4,TLR1/2 agonist;FSL-1,TLR2/6 agonist)处理后的信号分子激活情况(如磷酸化EGFR、磷酸化PTK2、NF-κB亚基p65);③双荧光素酶报告基因实验:检测PTK2-KO对NF-κB转录活性的影响(处理因素为Pam3CSK4或FSL-1)。结果解读:IP实验显示PTK2与EGFR存在直接相互作用,且PTK2过表达可增强EGFR的磷酸化水平(条带灰度值升高约2倍,P<0.01);TLR agonists处理后,对照细胞中PTK2磷酸化水平升高(如Pam3CSK4处理30分钟后升高约1.5倍),而PTK2-KO细胞中NF-κB转录活性显著降低(如FSL-1处理后,PTK2-KO细胞NF-κB活性较对照降低约50%,P<0.001),表明PTK2通过促进EGFR磷酸化及TLRs介导的NF-κB激活,调控两条信号通路的交叉对话。产品关联:实验所用关键试剂包括Flag抗体(如Sigma-Aldrich F1804)、Myc抗体(如Cell Signaling Technology 2276)、NF-κB双荧光素酶报告基因试剂盒(如Promega E846A)。
3.6 PTK2抑制剂治疗效果验证
实验目的是验证PTK2抑制剂Defactinib对EGFR或TLRs诱导肺癌进展的抑制作用。方法细节:①通过CCK-8法确定Defactinib对A549、H1299细胞的半数抑制浓度(IC₅₀);②3D肿瘤球形成实验:将野生型A549细胞接种于96孔板,培养2天后加入Defactinib(9.7μM)及EGFR/TLRs刺激因子(如EGF、Pam3CSK4、FSL-1),观察肿瘤球大小变化(n=5)。结果解读:Defactinib的IC₅₀在A549细胞中约为9.7μM;与vehicle处理组相比,Defactinib显著抑制EGF、Pam3CSK4或FSL-1诱导的肿瘤球形成(如EGF+Defactinib组肿瘤球大小较EGF组减少约40%,n=5,P<0.01),且对“EGF+TLR agonist”联合处理的抑制效果更显著(如Pam3CSK4+EGF+Defactinib组肿瘤球大小较联合处理组减少约60%,P<0.001)。产品关联:实验所用抑制剂为Defactinib(购自Selleck Chemicals,货号S7111);CCK-8试剂盒同前。
4. Biomarker 研究及发现成果解析
本研究定位的Biomarker为蛋白酪氨酸激酶2(PTK2),属于“基因表达类生物标志物”,其筛选与验证逻辑遵循“临床样本筛选→细胞功能验证→体内模型确认→治疗响应验证”的完整链条:首先通过42例NSCLC患者的肿瘤与正常组织微阵列数据,筛选出PTK2高表达与患者不良预后相关;接着通过细胞模型验证PTK2缺失抑制肿瘤进展;再通过动物模型确认PTK2缺失抑制体内肿瘤生长;最后通过抑制剂实验验证PTK2靶向抑制对EGFR/TLRs诱导肿瘤进展的抑制作用。
研究过程中,PTK2的来源为NSCLC患者的肿瘤组织及配对正常组织(临床样本)、肺癌细胞系(A549、H1299)及NSG小鼠异种移植肿瘤组织。验证方法包括:①临床样本的基因表达谱分析(ΔMag分层)与生存分析;②细胞水平的功能实验(增殖、迁移、3D肿瘤球);③动物水平的肿瘤生长与转移观察;④分子水平的相互作用实验(IP、WB)及转录活性检测(双荧光素酶报告基因)。特异性与敏感性数据方面,PTK2高表达(PTK2up)患者的总生存期显著短于低表达(PTK2down)患者(中位生存期18个月 vs 36个月,n=26 vs n=16,P<0.01);EGFR与PTK2共高表达(EGFRupPTK2up)患者的生存百分比显著低于共低表达患者(29% vs 86%,n=21 vs n=7,P<0.001)。
核心成果包括:①PTK2是EGFR或TLRs诱导NSCLC进展的潜在预后生物标志物——其高表达与NSCLC患者的不良预后密切相关,可作为判断EGFR或TLRs信号激活型NSCLC的预后指标;②PTK2通过调控EGFR与TLRs信号的交叉对话促进肿瘤进展,首次揭示了“PTK2-EGFR-TLRs”信号网络在NSCLC中的作用机制;③PTK2是EGFR或TLRs诱导NSCLC的潜在治疗靶点——其抑制剂Defactinib可有效抑制EGFR/TLRs诱导的肿瘤进展,为NSCLC的精准治疗提供了新的候选靶点。