1. 领域背景与文献引入
文献英文标题:Transcriptomic changes and gene fusions during the progression from Barrett’s esophagus to esophageal adenocarcinoma;发表期刊:Biomarker Research;影响因子:未公开;研究领域:食管腺癌发生机制(Barrett食管向食管腺癌进展的分子机制)。
食管腺癌(EAC)是全球发病率增长最快的恶性肿瘤之一,近几十年发病率激增600%,但其5年生存率仅约15%,预后极差。Barrett食管(BE)是EAC的明确前驱病变,约2%的人群受累,可使EAC发病风险升高40倍。然而,尽管BE与EAC的临床关联已明确,两者之间的转录组动态变化、关键分子通路及基因融合事件仍不清楚——现有研究多聚焦于单一分子类型(如miRNA),缺乏对BE到EAC进展的全面转录组解析,也鲜有研究关注基因融合的组织特异性。这些知识空白严重阻碍了EAC的早期诊断和靶向治疗策略的开发。
针对上述问题,本研究通过对8例患者的BE和EAC组织进行全面的RNA测序(RNA-seq),结合差异基因表达分析、通路网络解析、基因融合预测及多维度验证(单细胞RNA-seq、免疫荧光、RT-PCR、纳米孔测序),旨在揭示BE向EAC进展中的关键mRNA特征和基因融合事件,为EAC的早期诊断、预防及治疗提供新的生物标志物和分子靶点。
2. 文献综述解析
文献综述部分围绕“EAC的临床困境”“BE的前驱病变角色”“现有研究的局限性”三个核心维度展开评述。现有研究的关键结论包括:① EAC发病率高、生存率低,是消化系统肿瘤的重要挑战;② BE作为EAC的前驱病变,其癌变风险较正常人群高40倍,但分子机制未明;③ 此前研究虽报道了EAC的miRNA profiling等结果,但缺乏对BE到EAC进展的全面转录组分析,且样本多为非配对组织,难以揭示动态分子变化。
现有研究的技术方法优势在于:部分研究利用组学技术(如miRNA测序)筛选了EAC的生物标志物;局限性则体现在:样本量小、缺乏配对的BE与EAC组织、未整合基因融合分析及功能验证。本研究的创新点在于:采用配对的BE与EAC样本(n=8),整合RNA-seq、单细胞验证及功能实验(免疫荧光、基因融合验证),全面解析了BE到EAC的转录组变化及基因融合事件,弥补了现有研究在样本配对、多组学整合及功能验证上的不足,为BE到EAC的分子机制提供了更系统的证据。
3. 研究思路总结与详细解析
本研究的目标是揭示Barrett食管向食管腺癌进展过程中的转录组变化及基因融合事件;核心科学问题是BE到EAC进展中的关键分子改变(差异基因、通路及基因融合)及其在癌变中的作用;技术路线为“样本收集→RNA-seq测序→差异基因分析→通路与网络分析→基因融合预测→单细胞RNA-seq验证→免疫荧光验证→RT-PCR及纳米孔测序验证”,形成从组学筛选到功能验证的闭环。
3.1 样本收集与RNA测序
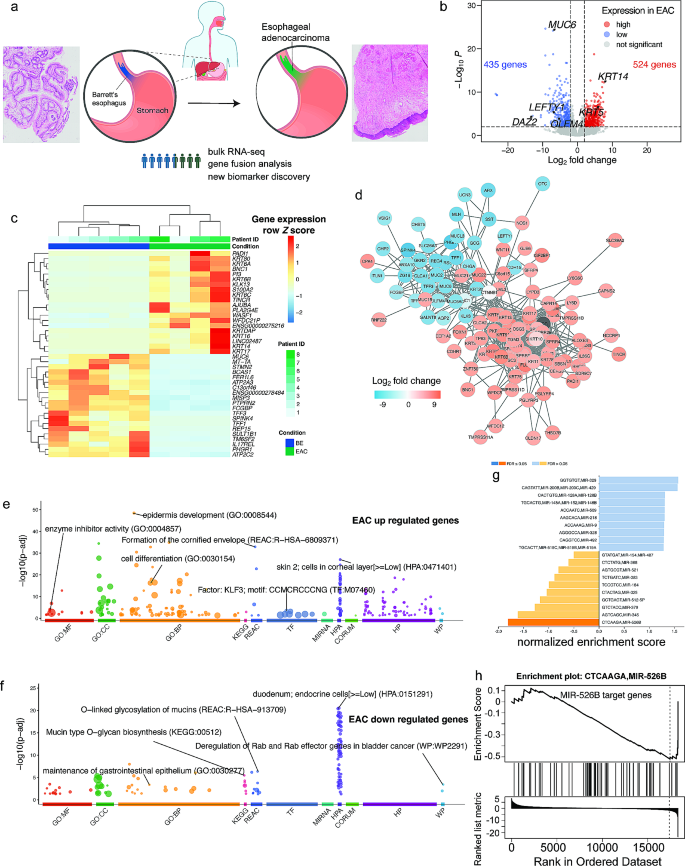
实验目的是获取BE和EAC组织的转录组数据,为后续差异分析奠定基础。方法细节为收集8例患者的BE和EAC配对组织,提取总RNA后进行RNA-seq(Illumina平台,具体测序参数未提及)。结果解读:通过差异基因表达分析(以log₂倍数变化(FC)和调整后P值为标准),鉴定出524个在EAC中显著上调的基因和435个显著下调的基因(图1b),其中角质蛋白家族(如KRT14)、黏蛋白基因(如MUC5B)为关键差异基因。实验所用关键产品:文献未提及具体实验产品,领域常规使用RNA提取试剂盒(如Qiagen RNeasy Mini Kit)、RNA-seq文库构建试剂盒(如Illumina TruSeq Stranded mRNA Library Prep Kit)。
3.2 差异基因与功能通路分析
实验目的是解析差异基因的功能及参与的信号通路,揭示BE到EAC的分子变化规律。方法细节为对差异基因进行基因本体(GO)、京都基因与基因组百科全书(KEGG)、Reactome等通路富集分析,同时通过基因集富集分析(GSEA)寻找调控差异基因的miRNA。结果解读:上调基因富集于“表皮发育”“细胞分化”等发育相关过程,且富集KLF3转录因子靶点(图1e),提示EAC细胞存在去分化趋势;下调基因富集于“黏蛋白O-糖基合成”“十二指肠内分泌细胞功能”(图1f),反映BE上皮功能的丢失。GSEA分析发现miR-526B的靶基因在EAC中显著下调(FDR显著),其靶基因包括DAZ1-4、ARX等,进一步支持去分化机制(图1g-h)。实验所用关键产品:文献未提及具体实验产品,领域常规使用通路分析软件(如DAVID、GSEA)。
3.3 基因融合预测与验证
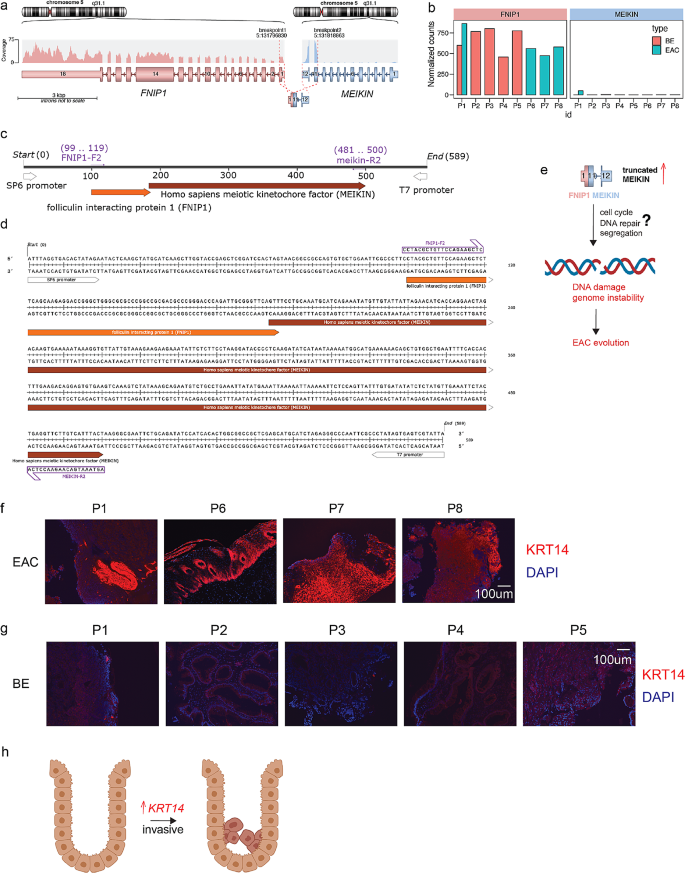
实验目的是鉴定BE到EAC进展中的基因融合事件,明确其组织特异性。方法细节为通过RNA-seq数据预测基因融合,对候选融合(如Patient 1的FNIP1-MEIKIN)采用RT-PCR扩增融合片段,并用纳米孔测序验证序列准确性,同时分析融合基因的表达水平。结果解读:多数基因融合为患者特异性,其中Patient 1的FNIP1-MEIKIN融合由FNIP1的启动子区域与MEIKIN的最后两个外显子融合而成,导致MEIKIN在EAC中高表达(图2a-d);该融合仅存在于EAC组织中,未在配对BE样本中检测到(图S4)。推测该融合通过激活减数分裂基因MEIKIN,导致染色体分离缺陷和基因组不稳定,进而促进癌症进展(图2e)。实验所用关键产品:文献未提及具体实验产品,领域常规使用RT-PCR试剂盒(如Thermo Fisher SuperScript IV One-Step RT-PCR System)、纳米孔测序仪器(如Oxford Nanopore MinION)。
3.4 KRT14蛋白水平的免疫荧光验证
实验目的是验证差异基因KRT14在蛋白水平的表达变化,明确其组织特异性。方法细节为对BE和EAC组织进行免疫荧光染色,使用KRT14特异性抗体标记,DAPI染核,观察荧光信号强度。结果解读:免疫荧光结果显示,EAC组织中KRT14的红色荧光信号显著强于BE组织(图2f-g),与RNA-seq的mRNA上调趋势一致。推测KRT14通过激活ROCK-1信号通路等机制,促进EAC细胞的侵袭和迁移(图2h),这与之前肺癌研究中KRT14的功能一致。实验所用关键产品:文献未提及具体实验产品,领域常规使用KRT14抗体(如Abcam Anti-KRT14 Antibody)、免疫荧光试剂盒(如Thermo Fisher Alexa Fluor 594 Conjugated Secondary Antibody)。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究筛选并验证了两类生物标志物:一是差异表达mRNA(如角质蛋白14,KRT14),二是基因融合事件(如FNIP1-MEIKIN)。筛选与验证逻辑为:通过RNA-seq筛选差异基因→单细胞RNA-seq验证表达趋势→免疫荧光验证蛋白水平(KRT14);基因融合通过RNA-seq预测→RT-PCR验证存在性→纳米孔测序验证序列→分析组织特异性(仅EAC存在)。
研究过程详述
KRT14的来源为8例患者的BE和EAC组织,验证方法为免疫荧光染色,结果显示EAC组织中KRT14的荧光强度显著高于BE(样本量n=8,文献未提供具体P值,但视觉差异明显);FNIP1-MEIKIN融合的来源为Patient 1的EAC组织,验证方法为RT-PCR和纳米孔测序,结果显示该融合仅存在于EAC中,未在配对BE中检测到(n=1,P值未明确),且MEIKIN的表达水平在EAC中显著升高(图2b)。
核心成果提炼
KRT14作为EAC的潜在预后或诊断生物标志物,其在EAC中的高表达与细胞侵袭、迁移能力相关(创新性:首次在BE到EAC进展中明确KRT14的蛋白水平变化及功能关联);FNIP1-MEIKIN融合为EAC特异性分子事件,可能通过激活MEIKIN导致基因组不稳定,进而驱动癌变(创新性:首次报道该融合在BE到EAC转化中的作用)。这些生物标志物不仅为EAC的早期诊断提供了新的分子靶点,也为理解BE向EAC的转化机制提供了关键线索,具有重要的临床应用价值和学术意义。