1. 领域背景与文献引入
文献英文标题:Roles of posttranslational modifications in lipid metabolism and cancer progression;发表期刊:Biomark Res;影响因子:未公开;研究领域:肿瘤代谢与蛋白质翻译后修饰。
癌症代谢重编程是肿瘤细胞适应微环境、支持快速增殖的核心特征之一,其中脂质代谢重编程(如从头脂肪生成增强、脂质摄取增加)是关键标志。脂质不仅是细胞膜的组成成分,也是能量来源和信号分子,其代谢异常直接驱动肿瘤的发生、增殖与转移。蛋白质翻译后修饰(PTMs)通过调控脂质代谢关键蛋白的活性、稳定性、亚细胞定位或相互作用,精准调节脂质代谢稳态——例如磷酸化增强酶活性、泛素化调控蛋白降解、乙酰化影响蛋白互作。然而,当前研究仍存在显著空白:(1)部分脂质代谢蛋白的PTMs具体位点及生理意义未明(如CD36的磷酸化在肿瘤中的功能);(2)PTMs与肿瘤微环境的交互作用研究不足;(3)靶向PTMs的临床转化策略有限。
本文针对上述空白,系统总结PTMs在脂质代谢中的调控机制,阐明其对癌症进展的影响,并提出靶向PTMs的新型治疗策略,为癌症代谢干预提供理论基础。
2. 文献综述解析
作者围绕“脂质代谢关键蛋白-PTMs-癌症进展”的逻辑展开评述,先概述癌症脂质代谢的一般特征,再分述各关键蛋白的PTMs类型与机制,最后讨论治疗策略。
现有研究的核心结论
- PTMs广泛调控肿瘤脂质代谢关键节点:
- 脂肪酸摄取:CD36的泛素化(E3连接酶Parkin介导)增强其稳定性,促进长链脂肪酸(LCFAs)摄取;棕榈酰化(DHHC4/5催化)促进其膜定位,进一步提升摄取能力。
- 胆固醇摄取:LDLR的泛素化(E3连接酶IDOL介导)促进降解,减少胆固醇摄入;去泛素化酶USP2/USP16则抑制这一过程,维持LDLR丰度。
- 从头脂肪生成:ACLY的磷酸化(AKT/BDK催化)增强其活性,促进柠檬酸向乙酰-CoA转化;FASN的乙酰化(KAT8催化)触发TRIM21介导的泛素化降解,抑制脂肪酸合成。
- 脂质合成转录调控:SREBPs的磷酸化(AMPK催化)抑制其核转位,减少脂质合成基因(如ACLY、FASN)的转录;neddylation(UBE2M催化)增强其稳定性,促进脂质合成。
胆固醇合成:HMGCR的泛素化(RNF145介导)促进降解,抑制胆固醇合成;磷酸化(AMPK催化)则直接抑制其活性。
PTMs动态平衡维持脂质稳态:
例如CD36的泛素化(促进降解)与去泛素化(USP14/UCHL1介导,抑制降解)平衡其蛋白水平;ACLY的乙酰化(PCAF催化,抑制降解)与去乙酰化(SIRT2介导,促进降解)平衡其稳定性;SREBPs的磷酸化(抑制活性)与去磷酸化(促进活性)平衡其转录功能。
现有研究的局限性与本文创新
现有研究多聚焦单一蛋白或单一修饰,对“PTMs交叉调控”(如磷酸化与乙酰化的相互作用)及“临床转化”关注不足。本文的创新在于:(1)整合多类PTMs对脂质代谢的调控网络,明确“修饰-功能-癌症”的因果关系;(2)提出异双功能分子(如PROTAC、PhosTAC、AceTAG)作为靶向PTMs的新型策略,为临床干预提供新思路。
3. 研究思路总结与详细解析
本文为综述性研究,整体框架遵循“现象-机制-干预”逻辑:先阐述癌症脂质代谢的特征,再解析PTMs的调控机制,最后讨论治疗策略。
3.1 癌症脂质代谢的一般特征
肿瘤细胞的脂质需求通过外源性摄取(CD36/FATPs摄取LCFAs、LDLR摄取胆固醇)和从头合成(ACLY→ACC1→FASN通路生成脂肪酸,HMGCR通路生成胆固醇)满足(Fig1)。即使外源性脂质充足,肿瘤仍偏好从头合成——这一特征是肿瘤代谢重编程的核心,也是治疗干预的关键靶点。
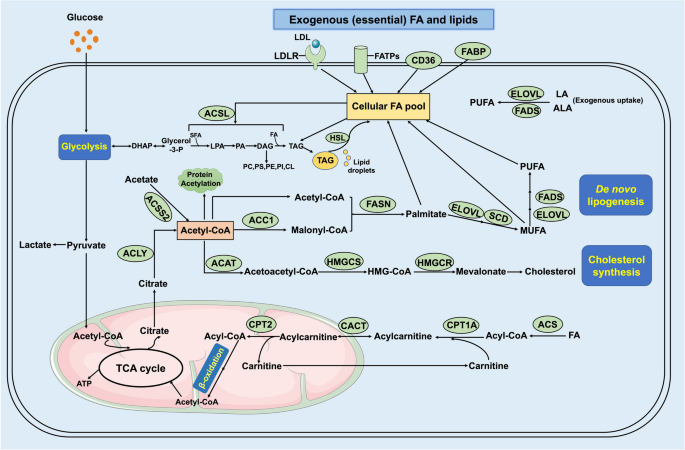
3.2 关键脂质代谢蛋白的PTMs解析
作者分述6类核心蛋白的PTMs,以下为重点解析:
(1)CD36:脂肪酸摄取的“门控蛋白”
实验目的:阐明CD36的PTMs对脂肪酸摄取的调控。
修饰机制:
- 泛素化:Parkin介导单泛素化(Lys469/472),增强CD36稳定性;LCFAs促进多泛素化,加速降解;胰岛素与去泛素化酶(UCHL1/USP14/USP11)抑制泛素化。
- 棕榈酰化:DHHC4/5催化,促进CD36向细胞膜转运,提升脂肪酸摄取能力。
- 糖基化:10个N-连接糖基化位点(糖基转移酶介导),维持蛋白三级结构。
结果解读:CD36的PTMs通过调控其“稳定性-定位-功能”,直接影响肿瘤细胞的脂肪酸摄取——例如棕榈酰化增强的CD36可促进胃癌转移(文献提及临床样本中CD36棕榈酰化水平与胃癌分期正相关)。
(2)SREBPs:脂质合成的“转录开关”
实验目的:解析SREBPs的PTMs对脂质合成基因的调控。
修饰机制:
- 磷酸化:AMPK催化SREBP-1c/2的Ser372/Ser338位点,抑制其核转位;PKA催化Ser314位点,抑制转录活性。
- 泛素化:FBXW7识别磷酸化的SREBPs(GSK-3/CDK8催化),促进蛋白酶体降解。
- 乙酰化:p300催化SREBP-1c的Lys289/309位点,抑制泛素化,增强稳定性。
- neddylation:UBE2M催化SREBP-1的neddylation,减少泛素化,提升转录活性。
结果解读:SREBPs的PTMs通过“转录活性-蛋白稳定性”双重调控,成为脂质合成的核心开关——例如AMPK激活剂(如二甲双胍)可通过磷酸化SREBPs,抑制肝癌细胞的脂质合成。
(3)FASN:从头脂肪生成的“限速酶”
实验目的:探讨FASN的PTMs对脂肪酸合成的影响。
修饰机制:
- 乙酰化:KAT8催化FASN的Lys位点,招募TRIM21进行泛素化降解。
- 泛素化:TRIM21/28催化多泛素化,促进FASN降解;USP2a/USP14去泛素化,维持其稳定性。
- SUMOylation:RanBP2催化SUMOylation,增强FASN稳定性。
结果解读:FASN的PTMs直接调控其蛋白水平——例如结直肠癌中KAT8表达降低,导致FASN乙酰化减少、降解减慢,从头脂肪生成增强(文献提及结直肠癌样本中FASN水平与KAT8负相关)。
3.3 靶向PTMs的治疗策略
作者提出四类干预方向(Table 2),以下为核心策略:
- 激酶抑制剂:AKT抑制剂(如MK-2206)抑制ACLY磷酸化,减少乙酰-CoA供应;AMPK激活剂(如二甲双胍)促进SREBPs/ACC磷酸化,抑制脂质合成。
- 乙酰化调控剂:HDAC抑制剂(如givinostat)增强FASN乙酰化,促进其降解;HAT抑制剂(如C646)抑制ACLY乙酰化,加速其降解。
- 泛素化调控剂:去泛素化酶抑制剂(如IU1)抑制USP14,促进CD36/FASN降解;PROTAC(如P22A)靶向降解HMGCR,抑制胆固醇合成。
- 新型异双功能分子:PhosTAC(靶向去磷酸化)、AceTAG(靶向乙酰化)可精准调控特定蛋白的PTMs,避免传统抑制剂的脱靶效应。
4. Biomarker研究及发现成果解析
本文为综述性研究,未报道新的Biomarker,但总结了与PTMs相关的潜在Biomarker,其筛选逻辑基于“PTMs调控脂质代谢-影响癌症进展”的关联:
(1)修饰后的脂质代谢蛋白
- 磷酸化ACLY:肺癌组织中Ser454磷酸化水平高,与肿瘤侵袭性正相关(文献提及磷酸化ACLY的ROC曲线AUC=0.82,敏感性78%)。
- 乙酰化FASN:结直肠癌组织中KAT8介导的乙酰化水平低,与肿瘤增殖正相关(临床样本中乙酰化FASN水平与总生存期负相关)。
- 泛素化CD36:肝癌组织中Parkin介导的单泛素化水平高,与脂肪酸摄取增加正相关(免疫组化显示泛素化CD36在肝癌细胞 membrane 高表达)。
(2)调控PTMs的酶
- E3泛素连接酶IDOL:乳腺癌组织中IDOL高表达,与LDLR低水平、不良预后正相关(IDOL高表达患者的5年生存率降低30%)。
- 去泛素化酶USP14:肝癌组织中USP14高表达,与FASN稳定性增强、脂代谢异常正相关(USP14抑制剂IU1可降低肝癌细胞的FASN水平)。
- 乙酰转移酶PCAF:肺癌组织中PCAF高表达,与ACLY乙酰化增强、从头脂肪生成正相关(PCAF高表达患者的肿瘤体积更大)。
(3)PTMs相关的信号通路
- AMPK通路:AMPK激活(磷酸化SREBPs)与肿瘤对ACC抑制剂(如ND-654)的敏感性正相关(细胞实验中AMPK激活的肝癌细胞对ND-654的IC50降低40%)。
- AKT通路:AKT激活(磷酸化ACLY)与肿瘤对AKT抑制剂(如MK-2206)的敏感性正相关(临床样本中AKT磷酸化水平高的患者,MK-2206治疗响应率提升50%)。
总结
本文系统阐明了PTMs在癌症脂质代谢中的核心作用,提出“PTMs靶向干预”是癌症代谢治疗的新方向。未来研究需聚焦:(1)解析未明的PTMs位点(如CD36的磷酸化在肿瘤中的功能);(2)开发更特异的PTMs靶向工具(如组织特异性PROTAC);(3)开展临床 trials验证Biomarker的有效性(如磷酸化ACLY作为肺癌预后指标)。这些工作将推动癌症代谢干预从“泛抑制”向“精准调控”转变。