1. 领域背景与文献引入
文献英文标题:Associations of single nucleotide polymorphisms with mucinous colorectal cancer: genome-wide common variant and gene-based rare variant analyses;发表期刊:Biomarker Research;影响因子:未公开;研究领域:结直肠癌分子标志物与遗传学
结直肠癌是全球范围内第三大常见癌症,对个体健康和医疗系统造成显著负担,2012年其发病率在发达国家更高,加拿大纽芬兰与拉布拉多地区的年龄标准化发病率和死亡率居全国首位。黏液性腺癌是结直肠癌的侵袭性亚型,占所有病例的5-15%,与非黏液性结直肠癌相比,该亚型患者发病年龄更轻、诊断时多处于晚期,且对系统治疗的反应性更差,但其遗传发病机制目前尚未明确。领域共识:现有结直肠癌遗传学研究已识别出多个致病基因,但针对黏液性亚型的研究多聚焦于MUC家族基因、BRAF突变、CpG岛甲基化表型等有限方向,且多采用候选基因策略,缺乏全基因组层面同时覆盖常见与罕见遗传变异的系统分析,无法全面揭示该亚型的遗传易感基础。本研究正是针对这一研究空白,旨在通过全基因组关联分析识别与黏液性结直肠癌表型相关的常见和罕见单核苷酸多态性(SNP),为解析其生物学机制、开发靶向治疗手段提供学术依据。
2. 文献综述解析
作者对领域内现有研究按研究策略(候选基因研究vs全基因组研究)和变异类型(常见变异vs罕见变异)进行分类评述。现有研究的关键结论包括:已证实MUC家族基因异常表达、BRAF突变、CpG岛甲基化表型与黏液性结直肠癌相关,部分候选基因研究揭示了该亚型与非黏液性亚型的分子差异;技术方法优势方面,候选基因研究针对性强,可快速验证已知通路与疾病的关联,全基因组研究则能无偏倚地筛选潜在易感位点;但现有研究存在明显局限性,如候选基因研究仅覆盖有限基因,样本量较小且缺乏临床验证的系统性,全基因组研究多聚焦常见变异,未充分分析罕见变异的贡献,且针对黏液性结直肠亚型的全基因组研究尚未见报道。本研究的创新价值在于,首次采用全基因组SNP数据同时分析常见和罕见变异与黏液性结直肠癌表型的关联,弥补了现有研究仅关注单一变异类型或候选基因的不足,为该亚型的遗传机制研究提供了全新的研究范式,有望识别出未被报道的易感位点与通路。
3. 研究思路总结与详细解析
本研究的整体框架为:以“识别黏液性结直肠癌的遗传易感变异”为核心目标,围绕“黏液性结直肠癌的遗传基础尚不明确”这一科学问题,构建“队列筛选→基因型数据质控→常见SNP关联分析→罕见变异区域分析→生物信息学功能验证”的闭环技术路线,通过多维度统计分析与功能注释,揭示与黏液性表型相关的遗传位点。
3.1 研究队列与基因型数据准备
实验目的是构建符合严格质量控制标准的结直肠癌患者队列及全基因组SNP数据集,为后续关联分析提供可靠基础。方法细节为:选取纽芬兰结直肠癌登记库中1999-2003年招募的505名白人患者,采用Illumina Omni1-Quad人类SNP基因分型平台进行全基因组SNP分型,利用PLINK v.1.07软件进行质量控制,排除不符合哈迪-温伯格平衡、缺失值>5%的SNP,以及性别信息不一致、重复样本、非白人血统、存在一级至三级亲属关系的患者,最终保留729373个常见SNP(次要等位基因频率MAF≥0.05)和275645个罕见SNP(MAF<0.05)。结果解读为:成功获得了高质量的研究队列和基因型数据集,确保了后续关联分析的准确性与可靠性。产品关联:实验所用关键产品:Illumina Omni1-Quad人类SNP基因分型平台,PLINK v.1.07软件。
3.2 常见SNP关联分析
实验目的是识别与黏液性结直肠癌表型相关的常见SNP,并验证其对表型的区分能力。方法细节为:首先采用单因素逻辑回归分析四种遗传模型(加性、共显性、显性、隐性),筛选出与黏液性表型关联的SNP;随后通过多因素逻辑回归调整基线变量(性别、诊断年龄、肿瘤分期、肿瘤位置、肿瘤分级),利用Akaike信息准则(AIC)选择每个SNP最合理的遗传模型,最终确定Top10关联SNP;进一步通过ROC曲线分析评估这些SNP对黏液性与非黏液性表型的区分能力,对比仅含基线变量、仅含Top SNP、同时含基线变量与Top SNP的三种模型的ROC曲线下面积(AUC)。结果解读为:未发现达到传统全基因组显著性水平(P<5×10^-8)的关联,但Top10 SNP显著提升了模型的区分能力,仅含基线特征的模型AUC为0.703(95%CI:0.634-0.773),加入Top SNP后AUC提升至0.916(95%CI:0.873-0.960),且两种模型的置信区间无重叠,表明区分能力的提升具有统计学显著性。
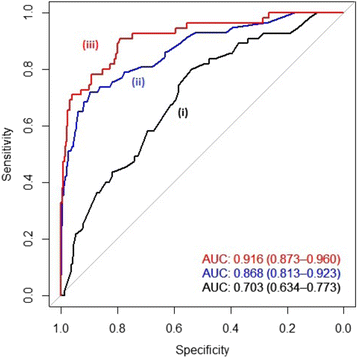
产品关联:实验所用关键产品:R v.3.1.3软件、pROC包。
3.3 罕见SNP关联分析
实验目的是识别与黏液性结直肠癌表型相关的罕见变异基因区域,弥补常见变异分析的局限性。方法细节为:采用基于基因区域的SKAT-O多标记检验,将每个基因上下游5kb序列纳入分析区域,利用Ensembl数据库的biomaRt工具获取基因位置信息,从患者全基因组数据中提取对应区域的SNP,在调整基线变量后分析基因区域与黏液性表型的关联。结果解读为:共分析29966个基因区域,发现多个潜在含因果罕见变异的区域(P<10^-4),其中SEC24B、SEC24B-AS1、CCDC109B区域位于4号染色体相近位置,其他区域分布于不同基因组位置,这些区域的基因多编码转运蛋白、调控RNA等分子,可能参与黏液产生的相关通路。产品关联:实验所用关键产品:SKAT-O分析工具、Ensembl数据库biomaRt工具。
3.4 生物信息学功能验证
实验目的是解析识别出的SNP与基因区域的潜在调控功能,揭示其与黏液性结直肠癌的生物学关联。方法细节为:利用RegulomeDB数据库分析SNP的调控特征,检索Ensembl数据库获取基因的功能注释信息。结果解读为:发现常见SNP中的kgp10457679(rs10819474)为表达数量性状位点(eQTL)、转录因子结合位点及DNA酶峰位点,可能调控肿瘤抑制基因PPP2R4的表达,该基因已被证实与结直肠癌患者生存相关;罕见变异区域中的LINC00596基因内rs8005541为eQTL,可能调控DHRS4和DHRS4L2基因的表达,这些基因此前未与黏液性结直肠癌关联,为后续机制研究提供了新方向。产品关联:实验所用关键产品:RegulomeDB数据库、Ensembl数据库biomaRt工具。
4. Biomarker研究及发现成果解析
本研究识别的Biomarker为与黏液性结直肠癌表型相关的常见SNP和罕见变异基因区域,筛选与验证逻辑为:全基因组SNP分型与质控→常见SNP的多因素逻辑回归分析+ROC曲线模型验证→罕见变异的基因区域SKAT-O分析→生物信息学功能注释。
研究过程详述:Biomarker来源为505名结直肠癌患者的外周血基因组DNA,常见SNP的验证采用单因素/多因素逻辑回归分析四种遗传模型,通过ROC曲线评估其对黏液性与非黏液性表型的区分能力,结果显示加入Top SNP后模型AUC从0.703提升至0.916(95%CI:0.873-0.960);罕见变异的验证采用SKAT-O多标记检验,识别出的关联基因区域显著性P<10^-4,每个区域包含5-10个罕见变异。
核心成果提炼:首次在全基因组层面同时分析常见和罕见SNP与黏液性结直肠癌的关联,识别出7个独立的常见SNP(排除高连锁不平衡后)和多个罕见变异基因区域,这些SNP涉及的基因参与DNA复制(CCDC141)、转录调控(ZBTB20)、分子转运(SLC22A16、SLC35F1)等生物学过程,为黏液性结直肠癌的遗传机制提供了新的线索;创新性在于突破了现有研究仅关注单一变异类型的局限,系统解析了常见与罕见变异的联合贡献;统计学结果方面,常见SNP模型区分能力的提升具有统计学显著性(置信区间无重叠),罕见变异区域的关联显著性P<10^-4(n=505)。这些Biomarker一旦在独立队列中得到验证,不仅能深化对黏液性结直肠癌生物学基础的理解,还可能为开发该亚型的早期诊断标志物与靶向治疗靶点提供依据。