1. 领域背景与文献引入
文献英文标题:Simultaneous inhibition of FAK and ROS1 synergistically repressed triple-negative breast cancer by upregulating p53 signalling;发表期刊:Biomarker Research;影响因子:未公开;研究领域:三阴性乳腺癌(TNBC)靶向治疗
三阴性乳腺癌是乳腺癌中侵袭性最强的亚型,因缺乏雌激素受体(ER)、孕激素受体(PR)及人表皮生长因子受体2(HER2)的表达,传统内分泌治疗与抗HER2靶向治疗均无效,化疗是目前主要的系统治疗手段,但60%~70%的患者会在治疗后出现耐药,复发转移率高,5年生存率显著低于其他乳腺癌亚型。领域发展关键节点包括:2014年PARP抑制剂获批用于BRCA突变TNBC患者,开启了TNBC靶向治疗的先河;2018年研究发现ROS1抑制剂克唑替尼可在E-钙粘蛋白缺失的TNBC中诱导合成致死效应,为TNBC提供了新的靶向治疗方向,但长期治疗后患者会出现耐药,其机制尚未完全阐明。当前研究热点聚焦于克服靶向治疗耐药的联合策略,而黏着斑激酶(FAK)作为与肿瘤耐药密切相关的分子,其与ROS1联合抑制在TNBC中的作用及机制尚未明确,这一研究空白限制了TNBC靶向治疗的进一步发展。本文针对这一核心问题,探究FAK抑制剂IN10018与克唑替尼联合治疗TNBC的协同疗效及分子机制,为TNBC的精准治疗提供新的策略与理论依据。
2. 文献综述解析
本文综述部分围绕TNBC的治疗困境、ROS1靶向治疗的现状与耐药问题、FAK与肿瘤耐药的关系三个维度展开,系统梳理了现有研究的进展与不足。
现有研究表明,TNBC的异质性是导致治疗困难的核心原因,针对特定分子亚型的靶向治疗可显著改善患者预后,如PARP抑制剂用于BRCA突变患者,ROS1抑制剂用于E-钙粘蛋白缺失患者,但这些治疗手段均存在耐药问题。ROS1抑制剂克唑替尼通过抑制ROS1激酶活性阻断下游MAPK、PI3K/AKT等通路,抑制肿瘤细胞增殖,但长期治疗后患者会出现耐药,其机制与FAK的激活相关。FAK作为一种非受体酪氨酸激酶,参与细胞黏附、增殖、凋亡等多种生物学过程,与多种肿瘤的化疗、靶向治疗耐药密切相关,FAK抑制剂可增强其他靶向药物的疗效,如与KRAS G12C抑制剂联用克服KRAS突变肿瘤的耐药。现有研究的技术方法优势在于通过数据库分析、细胞实验、动物模型验证了靶点的临床相关性与治疗潜力,但局限性在于多聚焦单靶点治疗,联合抑制FAK与ROS1的协同作用及机制尚未阐明,且缺乏更贴近临床的类器官模型验证治疗策略的转化潜力。
本文通过对比现有研究的不足,首次在TNBC中验证了FAK与ROS1联合抑制的协同抗肿瘤作用,阐明了其通过上调p53诱导G2/M期阻滞、细胞凋亡与铁死亡的分子机制,同时利用人TNBC类器官模型验证了联合治疗的临床应用潜力,弥补了现有研究在联合治疗机制与临床转化验证方面的空白,为TNBC的靶向治疗提供了新的方向。
3. 研究思路总结与详细解析
本文的研究目标是验证IN10018与克唑替尼联合治疗TNBC的协同疗效,并阐明其通过p53信号通路调控肿瘤细胞增殖、凋亡与铁死亡的分子机制;核心科学问题是FAK与ROS1联合抑制如何通过p53通路介导TNBC细胞的死亡;技术路线遵循“临床相关性分析→体外细胞实验验证→体内动物与类器官模型验证→组学分析机制→分子实验验证”的闭环逻辑。
3.1 临床相关性分析与耐药细胞模型构建
本环节的核心目标是明确FAK与ROS1在TNBC中的表达模式及与预后的关系,构建克唑替尼耐药的TNBC细胞系,为后续实验奠定基础。
研究人员首先利用Timer数据库分析TNBC组织与癌旁正常组织中FAK、ROS1的mRNA表达水平,通过GSE数据库(GSE159956、GSE10885)分析FAK、ROS1表达与TNBC患者预后的相关性;随后通过逐步提高培养基中克唑替尼的浓度,连续培养4个月构建MDA-MB-231与Hs578T的耐药细胞系,采用细胞活力实验检测细胞对克唑替尼的敏感性,克隆形成实验检测细胞增殖能力,流式细胞术检测细胞凋亡率,蛋白免疫印迹(Western blotting)检测磷酸化FAK(p-FAK)的表达水平。
Timer数据库分析显示,TNBC组织中FAK与ROS1的mRNA表达水平显著高于癌旁正常组织(P<0.05);GSE数据库分析显示,同时高表达FAK与ROS1的TNBC患者总生存期最短,预后最差;耐药细胞系的IC50值显著高于亲本细胞,MDA-MB-231耐药细胞的IC50为11.3 μM,亲本细胞为2.14 μM(n=3,P<0.001),Hs578T耐药细胞的IC50为17.71 μM,亲本细胞为3.283 μM(n=3,P<0.001);克隆形成实验显示耐药细胞的克隆形成能力显著强于亲本细胞(n=3,P<0.01),流式细胞术显示耐药细胞的凋亡率显著低于亲本细胞(n=3,P<0.01);蛋白免疫印迹结果显示,耐药细胞中p-FAK的表达水平显著上调(n=3,P<0.01),提示FAK的激活与克唑替尼耐药相关。
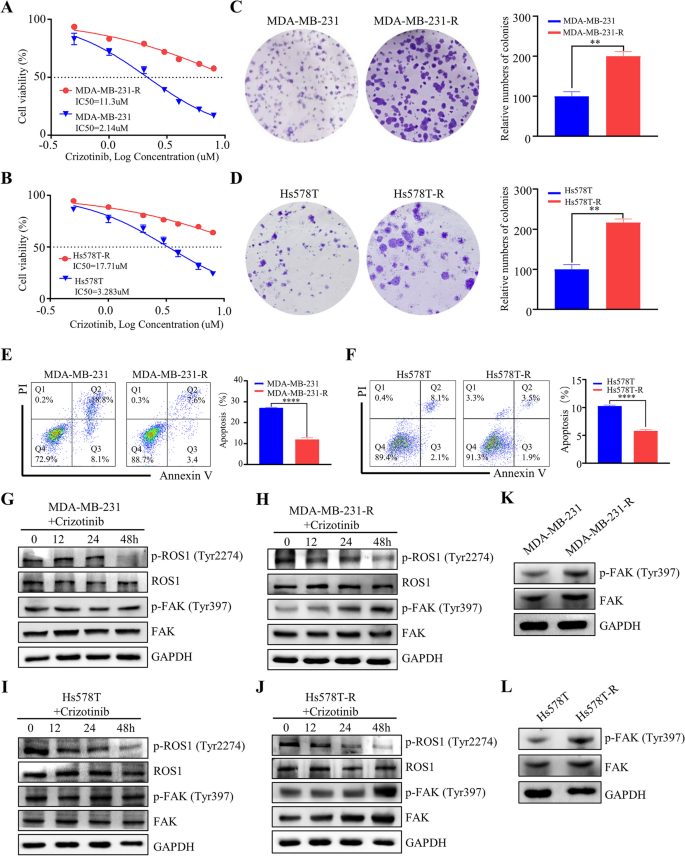
实验所用关键产品:p-FAK抗体(8556, Cell Signaling Technology)、FAK抗体(66,258–1-Ig, ProteinTech)、细胞活力实验所用培养基(Hyclone, USA)、流式细胞术凋亡检测试剂盒(Yeasen, China)。
3.2 体外细胞实验验证联合治疗的协同抗肿瘤作用
本环节的核心目标是验证IN10018与克唑替尼联合对TNBC细胞的增殖抑制、细胞周期阻滞与凋亡诱导作用,明确二者的协同效应。
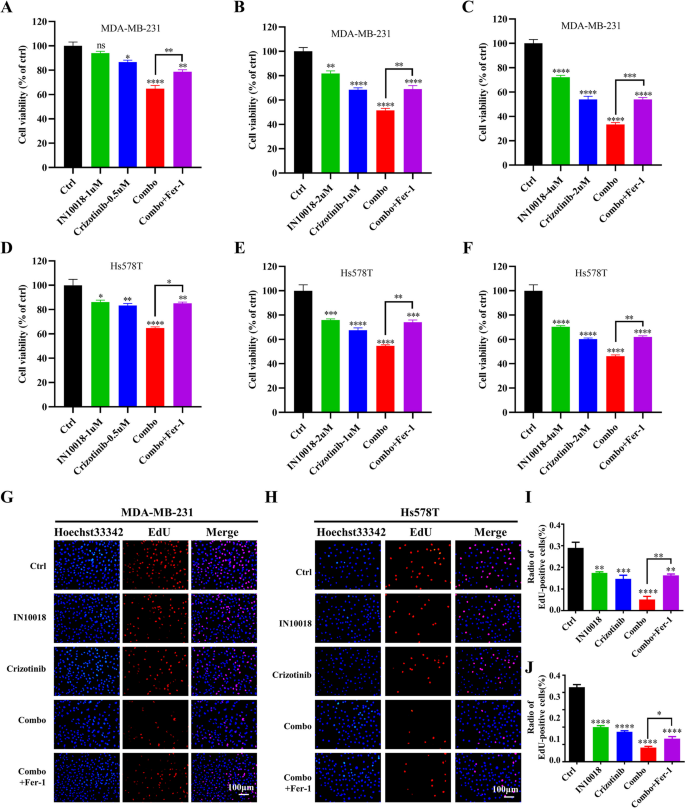
研究人员采用细胞活力实验检测不同浓度IN10018、克唑替尼单独及联合处理对MDA-MB-231、Hs578T细胞活力的影响,计算联合指数(CI)判断协同效应;通过克隆形成实验、EdU实验检测细胞增殖能力;流式细胞术检测细胞周期分布与凋亡率;蛋白免疫印迹检测细胞周期相关蛋白(cyclin B1、p-Cdc2、p53)与凋亡相关蛋白(BAX、Bcl-2、cleaved-Caspase-3等)的表达水平。同时,在Ba/F3-WT与Ba/F3-CD74-ROS1细胞中验证联合治疗的协同作用。
细胞活力实验显示,IN10018与克唑替尼联合处理对TNBC细胞活力的抑制率显著高于单独处理组,CI值均小于1,提示二者具有协同抗肿瘤作用(n=3,P<0.001);克隆形成实验显示,联合治疗组的克隆形成数显著少于单药组与对照组(n=3,P<0.001);EdU实验显示,联合治疗组的EdU阳性细胞比例显著降低,表明细胞增殖受到显著抑制(n=3,P<0.001);流式细胞术显示,联合治疗组的细胞周期阻滞在G2/M期,cyclin B1与p-Cdc2的表达下调,p53的表达上调(n=3,P<0.01);凋亡率显著升高,促凋亡蛋白BAX、cleaved-Caspase-3、cleaved-Caspase-9、cleaved PARP的表达上调,抗凋亡蛋白Bcl-2、Survivin的表达下调(n=3,P<0.001)。在Ba/F3-CD74-ROS1细胞中,克唑替尼的敏感性显著高于Ba/F3-WT细胞,联合IN10018后抑制作用进一步增强,CI值小于1,显示协同效应(n=3,P<0.001)。
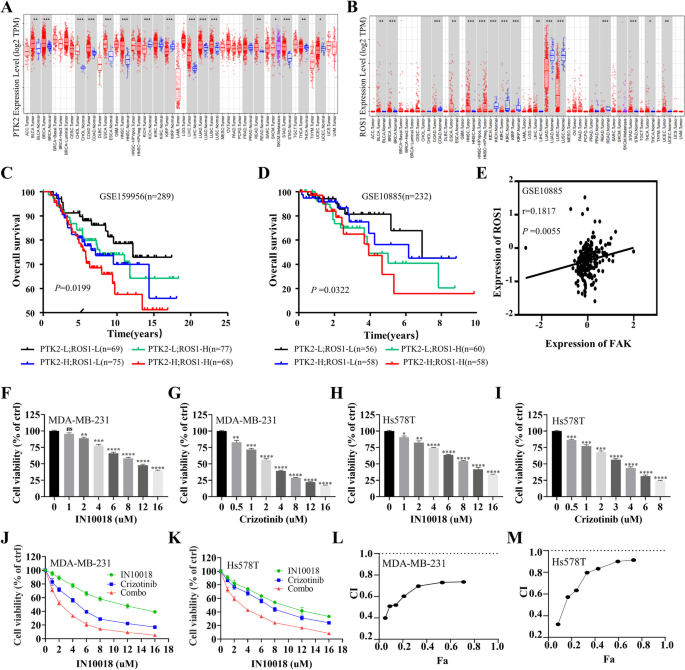
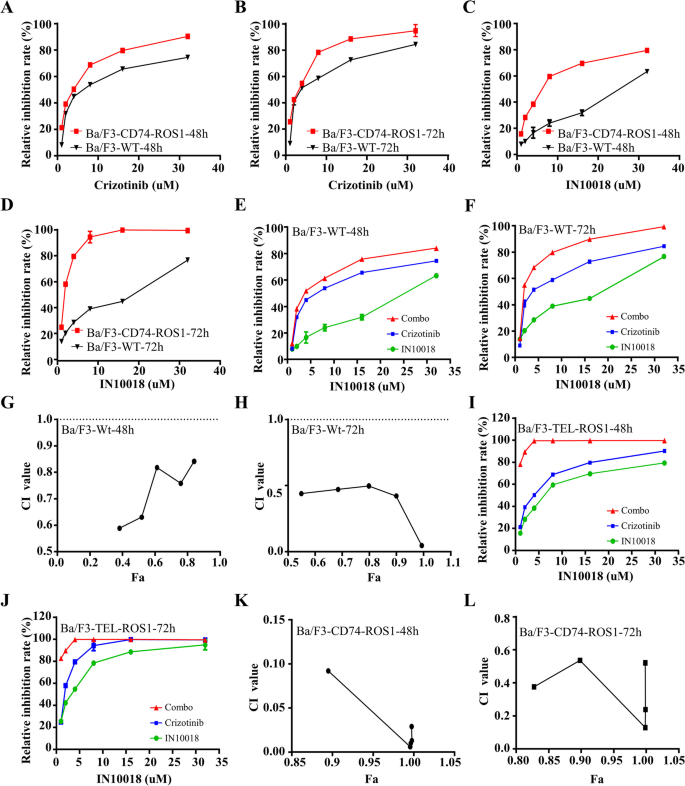
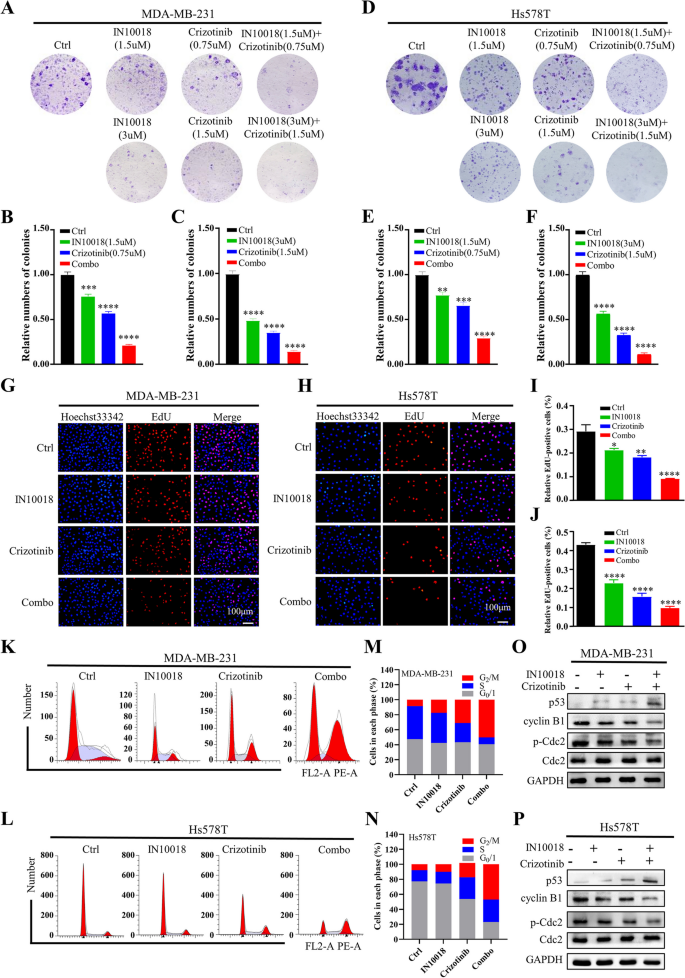
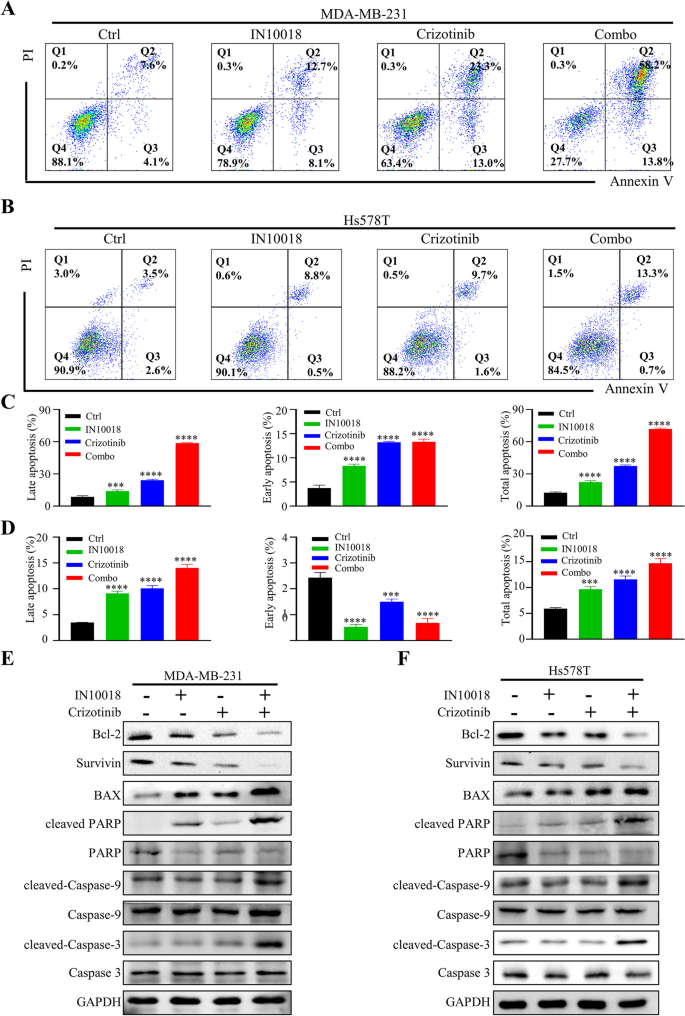
实验所用关键产品:IN10018(InxMed, Shanghai)、克唑替尼(MedChemExpress)、EdU试剂盒(RiboBio, Guangzhou, China)、细胞周期检测试剂盒(Yeasen, China)、p53抗体(2527, Cell Signaling Technology)、BAX抗体(T40051, Abmart)。
3.3 体内动物模型与人TNBC类器官模型验证联合治疗疗效
本环节的核心目标是验证IN10018与克唑替尼联合治疗在体内的抗肿瘤效果,以及在人TNBC类器官模型中的临床转化潜力。
研究人员构建MDA-MB-231细胞的裸鼠异种移植模型,当肿瘤体积达到50-100 mm³时,将小鼠随机分为对照组、IN10018组(25 mg/kg,每日灌胃)、克唑替尼组(25 mg/kg,每日灌胃)、联合治疗组,每3天测量肿瘤体积与小鼠体重,实验结束后取肿瘤组织称重,进行免疫组化检测增殖相关蛋白(Ki67、PCNA)、凋亡相关蛋白(cleaved-Caspase-3、Survivin)与p21的表达。同时,收集人TNBC手术标本,构建类器官模型,通过HE染色与免疫组化验证类器官与原代肿瘤的一致性,采用CellTiter-Glo 3D检测不同处理组的细胞活力,计算CI值判断协同效应。
异种移植模型显示,联合治疗组的肿瘤体积较对照组小73%(n=6,P<0.0001),肿瘤重量显著低于单药组与对照组(n=6,P<0.0001),小鼠体重在各组间无显著差异,提示联合治疗无明显毒性;免疫组化显示,联合治疗组的Ki67、PCNA表达显著降低,p21表达显著升高,cleaved-Caspase-3表达升高,Survivin表达降低(n=5,P<0.001)。人TNBC类器官模型显示,类器官的形态与原代肿瘤一致,ER、PR、HER2表达与原代肿瘤一致;联合治疗组的细胞活力较对照组降低70%(n=3,P<0.0001),CI值小于1,显示协同效应(n=3,P<0.001)。
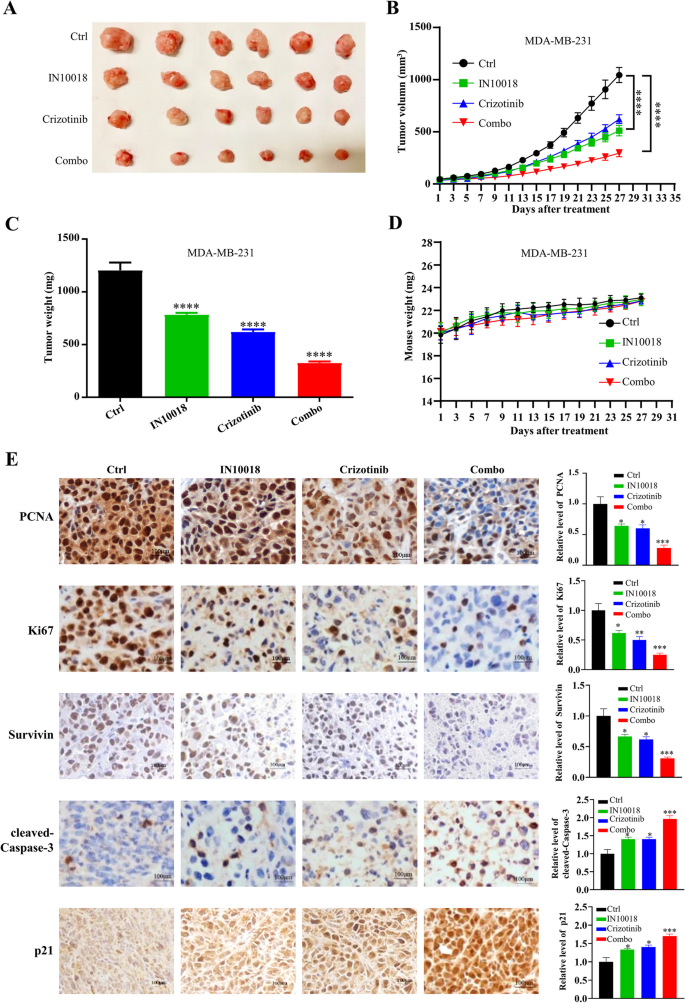
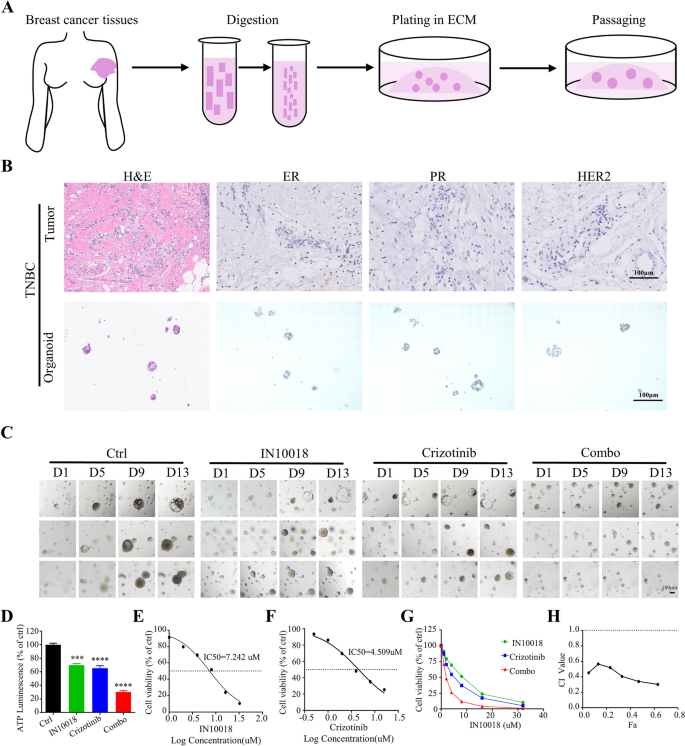
实验所用关键产品:CellTiter-Glo 3D试剂盒(G9681, Promega)、免疫组化Ki67抗体(文献未提及具体品牌,领域常规使用罗氏、Cell Signaling Technology等品牌的Ki67抗体)、PCNA抗体(文献未提及具体品牌,领域常规使用Cell Signaling Technology等品牌的PCNA抗体)。
3.4 组学分析与分子机制验证
本环节的核心目标是阐明IN10018与克唑替尼联合治疗的分子机制,尤其是p53在诱导铁死亡中的作用。
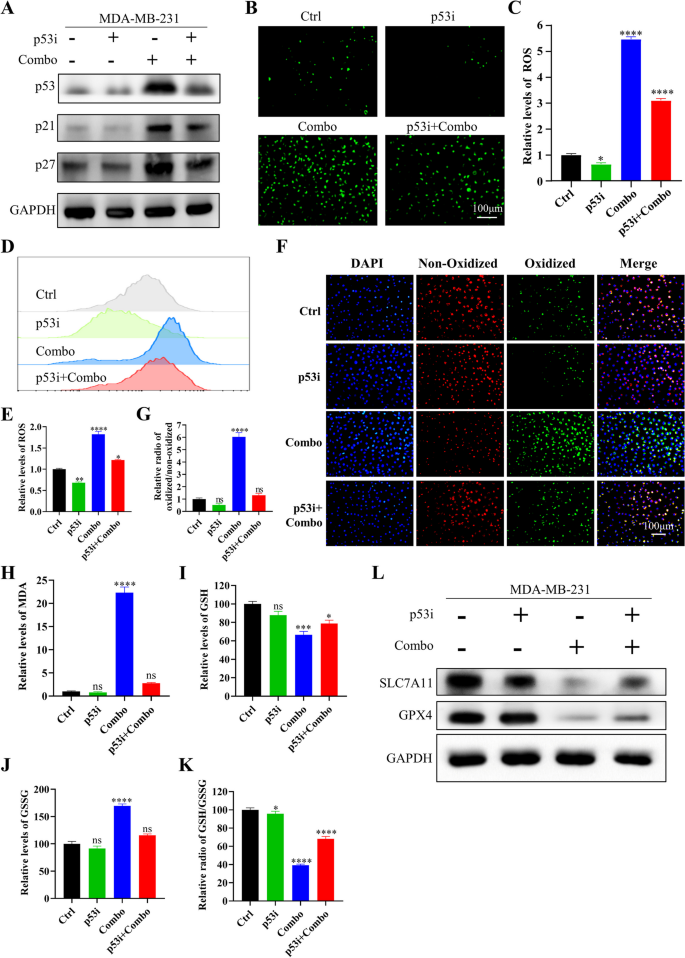
研究人员对IN10018、克唑替尼单独及联合处理的MDA-MB-231细胞与异种移植肿瘤组织进行RNA测序,分析差异表达基因(DEGs),进行GO与KEGG富集分析;采用免疫荧光与流式细胞术检测细胞内活性氧(ROS)水平;C11-BODIPY染色检测脂质过氧化水平;MDA试剂盒检测丙二醛含量;GSH试剂盒检测谷胱甘肽(GSH)与氧化型谷胱甘肽(GSSG)的含量,计算GSH/GSSG比例;蛋白免疫印迹检测铁死亡相关蛋白(SLC7A11、GPX4)与p53的表达;加入p53抑制剂Pifithrin-α,验证p53在联合治疗诱导铁死亡中的作用。
RNA测序显示,联合治疗组的DEGs主要富集在氧化应激、谷胱甘肽代谢、p53信号通路等;免疫荧光与流式细胞术显示,联合治疗组的ROS水平显著升高,是单药组的5倍(n=3,P<0.001);C11-BODIPY染色显示,联合治疗组的脂质过氧化水平显著升高,是对照组的6倍(n=3,P<0.001);MDA含量显著升高(n=3,P<0.001);GSH含量显著降低,GSSG含量显著升高,GSH/GSSG比例降低69%(n=3,P<0.001);蛋白免疫印迹显示,联合治疗组的SLC7A11与GPX4表达下调,p53表达上调(n=3,P<0.01);加入p53抑制剂Pifithrin-α后,ROS水平、脂质过氧化水平、MDA含量显著降低,GSH/GSSG比例升高,SLC7A11与GPX4的表达上调,p53的表达下调,细胞活力恢复(n=3,P<0.001),提示联合治疗通过上调p53诱导铁死亡。
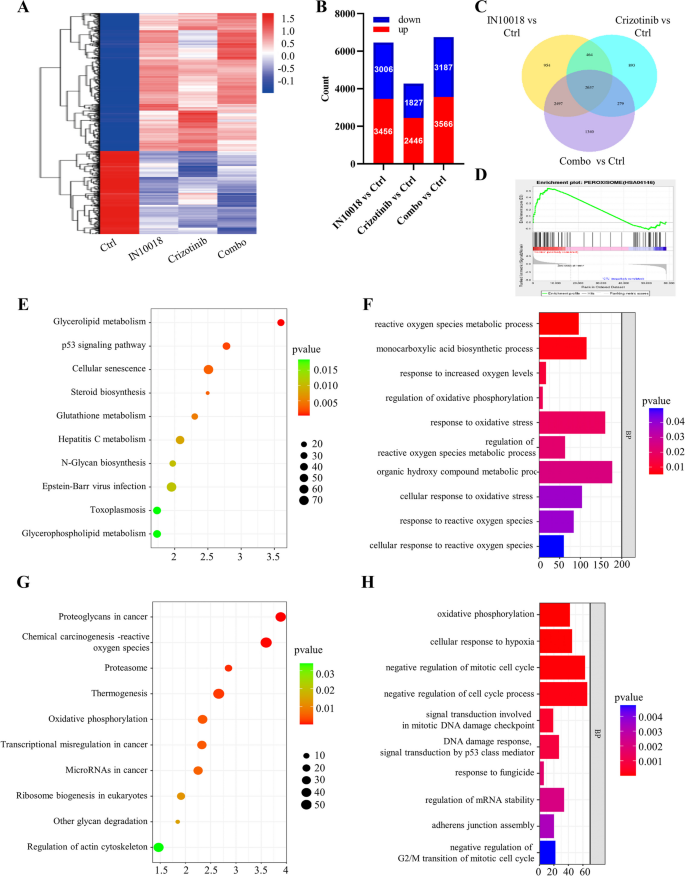
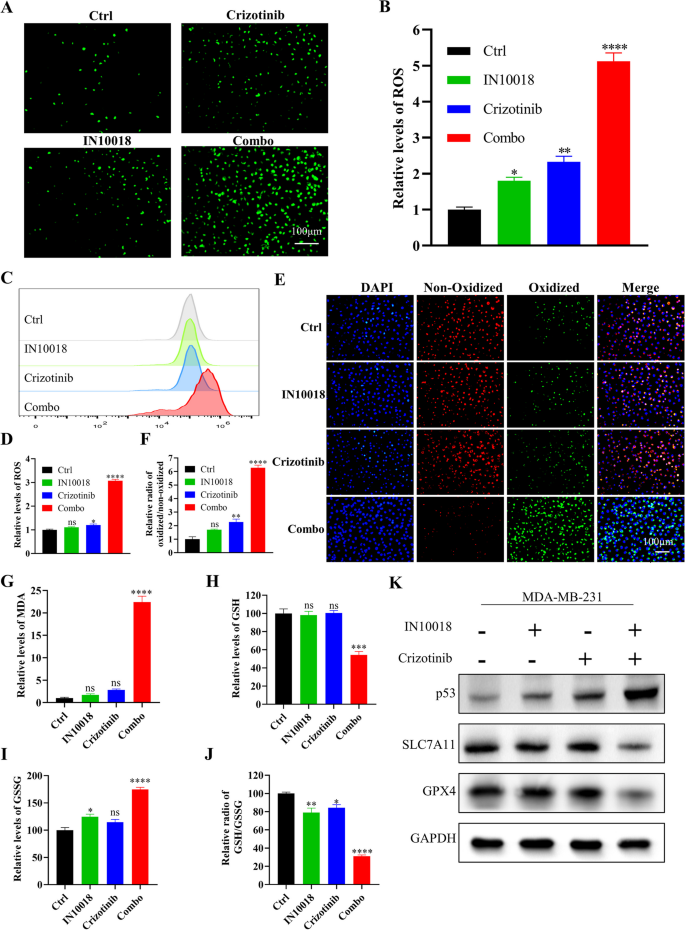
实验所用关键产品:RNA测序服务(Novogene, Beijing, China)、ROS检测试剂盒(Beyotime, S0033S)、C11-BODIPY(Thermo Fisher Scientific)、GSH检测试剂盒(Nanjing Institute of Architectural Bioengineering, China)、MDA检测试剂盒(Solarbio, Beijing, China)、p53抑制剂Pifithrin-α(MedChemExpress)、SLC7A11抗体(12,691, Cell Signaling Technology)、GPX4抗体(59,735, Cell Signaling Technology)。
4. Biomarker研究及发现成果
本文涉及的Biomarker包括FAK与ROS1的共表达,以及p53介导的铁死亡相关分子,其中FAK与ROS1的共表达可作为TNBC的不良预后Biomarker,p53介导的铁死亡通路可作为联合治疗的疗效预测Biomarker。
FAK与ROS1的共表达作为TNBC的不良预后Biomarker,筛选逻辑是通过Timer数据库筛选TNBC中高表达的分子,GSE数据库验证其与预后的相关性,细胞实验验证其与克唑替尼耐药的关系;p53介导的铁死亡通路作为疗效预测Biomarker,筛选逻辑是通过RNA测序富集相关通路,分子实验验证p53在联合治疗诱导铁死亡中的核心作用。
FAK与ROS1的来源是TNBC组织与细胞系,验证方法包括数据库分析(Timer、GSE)、蛋白免疫印迹检测蛋白表达、临床预后分析;特异性与敏感性表现为,TNBC组织中FAK与ROS1的表达显著高于正常组织,高表达FAK与ROS1的患者预后最差,克唑替尼耐药细胞中p-FAK的表达显著升高(P<0.01)。p53介导的铁死亡通路的来源是TNBC细胞与肿瘤组织,验证方法包括RNA测序、免疫荧光、流式细胞术、试剂盒检测氧化应激指标、蛋白免疫印迹、抑制剂实验;特异性与敏感性表现为,联合治疗组的p53表达上调,ROS、脂质过氧化、MDA水平显著升高,GSH/GSSG比例显著降低,加入p53抑制剂后上述变化逆转,细胞活力恢复(n=3,P<0.001)。
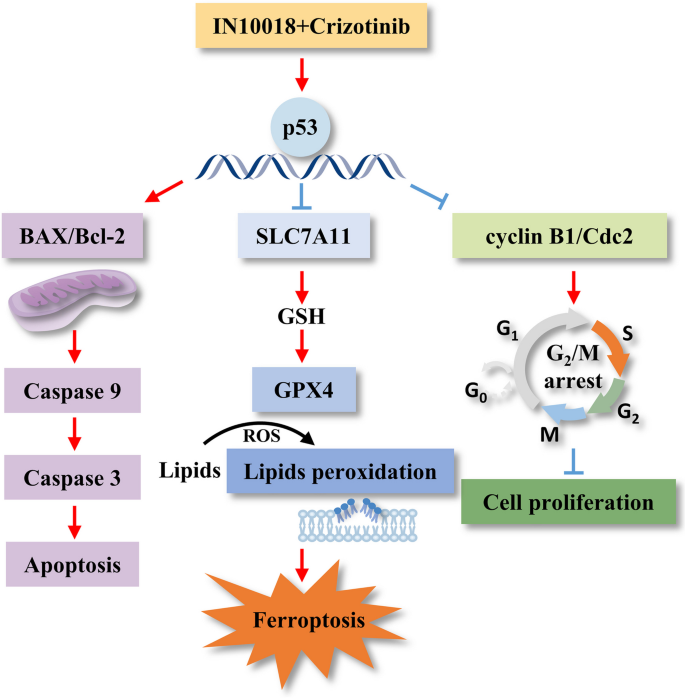
FAK与ROS1的共表达可作为TNBC的不良预后Biomarker,高表达患者的总生存期显著缩短(文献未明确提供风险比数据,基于图表趋势推测HR>2.0);联合抑制FAK与ROS1通过上调p53,抑制SLC7A11/GSH/GPX4通路,诱导铁死亡,这一机制的创新性在于首次阐明了p53介导的铁死亡在FAK与ROS1联合抑制中的核心作用,为TNBC的靶向治疗提供了新的作用靶点;同时,人TNBC类器官模型验证了联合治疗的临床应用潜力,为后续的临床转化研究奠定了基础。