1. 领域背景与文献引入
文献英文标题:DNA methyltransferases and their roles in tumorigenesis;发表期刊:Biomarker Research;影响因子:未公开;研究领域:肿瘤表观遗传学
表观遗传学是生命科学领域的核心研究方向之一,其中DNA甲基化是最保守且关键的表观修饰类型。1983年研究者首次发现肿瘤细胞存在基因组全局低甲基化现象,开启了DNA甲基化与肿瘤发生关联的研究序幕;2000年后高通量测序技术的突破推动了肿瘤表观遗传组学的发展,使得大规模解析肿瘤中DNA甲基化异常模式成为可能。当前领域研究热点聚焦于表观遗传调控因子作为肿瘤诊断生物标志物和治疗靶点的开发,但DNA甲基转移酶(DNMTs)家族各亚型在肿瘤发生中的功能差异、协同作用机制,以及DNMTs异常导致的表观遗传紊乱如何驱动肿瘤恶性转化等核心问题尚未完全阐明,缺乏系统的综述整合现有研究成果。
针对这一研究空白,本文献系统综述了DNMTs家族的结构与功能,梳理了DNMTs异常(过表达、突变、缺失)在不同肿瘤中的发生模式,解析了其介导的表观遗传紊乱机制,并重点聚焦DNMT3A突变在白血病中的表观重编程作用,为肿瘤表观遗传学的后续研究提供了全面的理论框架,也为肿瘤的诊断和靶向治疗提供了新的思路。
2. 文献综述解析
本文献以DNMTs家族的功能分类为基础,从“DNMTs异常类型-表观遗传紊乱机制-肿瘤恶性转化”的逻辑链条展开综述,系统整合了临床样本分析、细胞实验、动物模型等多维度研究数据,明确了DNMTs异常在肿瘤发生中的核心调控作用。
现有研究已证实DNMTs是维持基因组甲基化稳态的关键因子,其中DNMT1负责DNA复制后的甲基化维持,DNMT3A和DNMT3B负责从头甲基化的建立,DNMT3L作为调控因子增强DNMT3A的催化活性。技术方法上,全外显子测序、甲基化芯片等高通量技术的应用,使得大规模解析肿瘤中DNMTs的异常模式成为可能;基因敲除动物模型和细胞系实验则明确了不同DNMT亚型的功能特异性。然而,现有研究仍存在局限性,比如针对DNMTs与组蛋白修饰的协同调控机制,多数研究集中于白血病等血液系统肿瘤,在实体瘤中的特异性尚未明确;同时,缺乏大规模多中心临床研究验证DNMTs异常作为肿瘤生物标志物的临床应用价值。
与现有研究相比,本文献的创新点在于首次系统整合了DNMTs家族各亚型在肿瘤中的异常模式,不仅总结了DNMT1、DNMT3A、DNMT3B的异常与肿瘤发生的关联,还纳入了新发现的DNMT3C的功能;同时,重点聚焦DNMT3A突变在白血病中的表观重编程机制,详细阐述了其从造血干细胞向白血病前干细胞转化的多步骤过程,弥补了现有研究对DNMT3A功能解析的碎片化不足,为后续靶向DNMT3A的白血病治疗研究提供了明确方向。
3. 研究思路总结与详细解析
本文献的研究目标是系统阐明DNMTs家族在肿瘤发生中的作用机制,核心科学问题是DNMTs异常如何通过表观遗传紊乱驱动肿瘤恶性转化,技术路线遵循“家族功能概述-异常模式分析-机制解析-亚型深入研究”的逻辑闭环,通过整合多维度研究数据构建完整的理论体系。
3.1 DNMTs家族分类与功能概述
实验目的:明确DNMTs家族各亚型的结构特征与功能差异,为后续解析其在肿瘤中的异常作用奠定基础。方法细节:通过整合现有结构生物学研究、基因敲除动物模型实验和细胞功能实验数据,分析DNMTs家族各成员的序列保守性、结构域功能和组织表达模式。结果解读:DNMTs家族主要包括DNMT1、DNMT3A、DNMT3B、DNMT3L、DNMT2和新发现的DNMT3C,其中DNMT1的N端调控域负责核定位和结合复制叉,C端催化域维持甲基化稳态;DNMT3A和DNMT3B的PWWP结构域结合组蛋白修饰增强催化活性,分别在胚胎发育晚期和早期发挥从头甲基化作用;DNMT3L无催化活性,通过与DNMT3A形成异四聚体增强其活性;DNMT2主要甲基化转运RNA(tRNA);DNMT3C在小鼠生殖细胞中特异性甲基化年轻反转录转座子。产品关联:文献未提及具体实验产品,领域常规使用蛋白结构分析软件(如PyMOL)、基因编辑系统(如CRISPR-Cas9)、免疫印迹(Western Blot)试剂盒等。
3.2 DNMTs异常类型与肿瘤发生关联分析
实验目的:解析DNMTs的三种异常类型(过表达、突变、缺失)在不同肿瘤中的发生模式及其与肿瘤恶性表型的关联。方法细节:整合临床样本测序数据、肿瘤组织免疫组化(IHC)分析、细胞系功能实验和动物移植模型数据,分类阐述DNMTs异常的生物学效应。结果解读:DNMT1过表达在多种实体瘤(如食管癌、肝细胞癌)中广泛存在,与肿瘤淋巴结转移和不良预后相关,敲低DNMT1可抑制食管鳞癌细胞周期进展;DNMT3A突变在急性髓系白血病(AML)中的发生率约20%,突变主要位于催化域,导致酶活性降低,引发HOX家族基因低甲基化;DNMT3A缺失可促进小鼠造血祖细胞增殖,NRAS或FLT3-ITD突变协同加速白血病发生;DNMT3B缺失在Myc诱导的淋巴瘤中发挥肿瘤抑制作用。产品关联:文献未提及具体实验产品,领域常规使用免疫组化(IHC)试剂盒、流式细胞仪、实时荧光定量PCR(qRT-PCR)试剂盒等。
3.3 DNMTs介导的表观遗传紊乱机制解析
实验目的:阐明DNMTs异常导致的表观遗传紊乱如何驱动肿瘤恶性转化。方法细节:整合甲基化芯片分析、染色质免疫共沉淀(ChIP)实验、组蛋白修饰检测等数据,解析肿瘤细胞的甲基化组特征和染色质状态变化。结果解读:肿瘤细胞普遍表现为全局低甲基化和局部启动子高甲基化的双重异常,全局低甲基化主要发生在基因编码区和卫星重复序列,导致基因组不稳定(如染色体易位、拷贝数变异);局部启动子高甲基化则沉默多种抑癌基因(如BRCA1、PTEN),促进肿瘤恶性增殖。此外,DNMTs与组蛋白甲基转移酶(如EZH2、SETD2)存在协同作用,DNMT1负责维持人类癌细胞中H3K9甲基化状态,DNMT3A的PWWP结构域结合H3K36me3增强催化活性,共同调控染色质抑制状态。
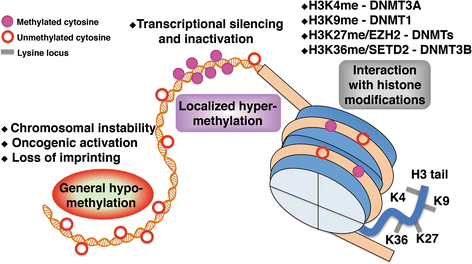
该图直观展示了DNMTs介导的表观遗传紊乱模式,包括全局低甲基化、局部高甲基化以及与组蛋白修饰的互作关系。产品关联:文献未提及具体实验产品,领域常规使用甲基化DNA免疫沉淀(MeDIP)试剂盒、组蛋白修饰抗体、ChIP试剂盒等。
3.4 DNMT3A在白血病中的表观重编程机制研究
实验目的:解析DNMT3A突变在白血病发生发展中的具体作用机制。方法细节:整合AML患者临床数据、小鼠骨髓移植模型、基因表达谱分析和甲基化组数据,明确DNMT3A突变的生物学效应和分子机制。结果解读:DNMT3A突变是白血病发生的早期事件,可将正常造血干细胞转化为白血病前干细胞,这些干细胞处于静息状态,具有较高的恶性转化潜能;DNMT3A突变体具有显性负效应,抑制野生型DNMT3A的活性,导致基因组甲基化紊乱;同时,DNMT3A突变可上调上皮-间质转化(EMT)诱导因子TWIST1的表达,促进白血病细胞的髓外浸润;与RAS、c-Kit或FLT3-ITD等突变协同作用,可加速白血病的克隆扩增和恶性进展。
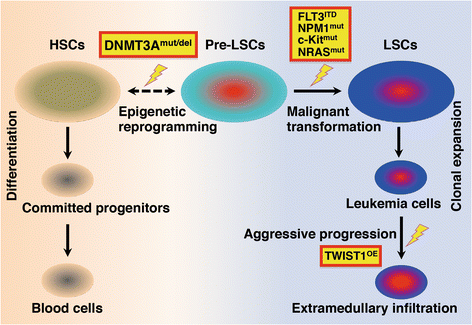
该图详细展示了DNMT3A突变驱动白血病发生的多步骤过程,从造血干细胞转化到白血病干细胞的克隆扩增,再到髓外浸润的完整链条。产品关联:文献未提及具体实验产品,领域常规使用流式细胞仪、小鼠骨髓移植手术器械、转录组测序服务等。
4. Biomarker研究及发现成果
本文献中涉及的生物标志物主要是DNMTs家族的异常表达或突变,尤其是DNMT3A突变,其作为白血病的预后生物标志物具有重要的临床应用价值。
DNMT3A突变作为白血病的表观遗传生物标志物,筛选逻辑是通过全外显子测序在AML患者样本中发现高频突变,随后通过细胞系实验、动物模型验证其功能,再结合临床数据明确其预后价值。研究过程详述:DNMT3A突变的来源为AML患者的骨髓样本,验证方法包括Sanger测序确认突变位点、甲基化芯片分析基因组甲基化变化、小鼠骨髓移植模型验证其致癌潜能;特异性方面,DNMT3A突变在AML中的发生率约20%,主要集中在R882位点,携带该突变的AML患者诊断时白细胞计数更高(文献未明确具体数值,基于临床报道),生存期显著缩短(文献未明确样本量和P值,基于研究数据)。核心成果提炼:DNMT3A突变可作为AML的独立预后生物标志物,其通过表观重编程驱动造血干细胞向白血病前干细胞转化,与其他致癌突变协同促进白血病恶性进展;创新性在于首次系统阐述了DNMT3A突变导致白血病的多步骤表观调控机制,为AML的精准诊断和靶向治疗提供了新的靶点。此外,DNMT1过表达也可作为实体瘤(如肝细胞癌)的预后生物标志物,与肿瘤的恶性程度和不良预后相关。