1. 领域背景与文献引入
文献英文标题:SMAD1 as a biomarker and potential therapeutic target in drug-resistant multiple myeloma;发表期刊:Biomarker Research;影响因子:未公开;研究领域:多发性骨髓瘤(MM)耐药机制与生物标志物研究。
多发性骨髓瘤是一种以骨髓中恶性浆细胞克隆增殖为特征的血液系统恶性肿瘤,尽管硼替佐米(BTZ)、地塞米松(DEX)等药物的应用显著改善了患者预后,但复发和化疗耐药仍是临床面临的核心挑战。目前,MM耐药的分子机制尚未完全阐明,缺乏有效的生物标志物预测耐药及复发,也亟需新的治疗靶点逆转耐药。SMAD1是TGF-β信号通路的核心介质,参与细胞生长、凋亡等过程,在实体瘤中被证实与肿瘤侵袭性及不良预后相关,但在MM中的作用尚未明确——其是否参与MM耐药调控、能否作为生物标志物或治疗靶点,均为未解决的关键问题。本研究针对这一空白,系统探索了SMAD1在MM耐药中的作用及机制,为耐药MM的诊断与治疗提供了新依据。
2. 文献综述解析
作者对现有研究的评述围绕三个核心维度展开:SMAD1在肿瘤中的功能(如实体瘤中促进侵袭、关联不良预后)、SMAD1与ID1的相互作用(ID1通过抑制p21/p27促进增殖)、NF-κB1/TNFAIP8轴在MM抗凋亡中的作用(TNFAIP8抑制caspase-8介导的凋亡,NF-κB1调控其表达)。现有研究的局限性在于:SMAD1在MM中的功能尚未报道;ID1在MM中的调控机制不清楚;SMAD1与NF-κB1的交互作用在MM中未被探索。
本研究的创新价值在于首次建立了SMAD1与MM耐药的因果关系:证明SMAD1通过两条独立轴调控MM耐药——① 上调ID1抑制p21/p27,促进细胞增殖;② 与NF-κB1相互调控,上调TNFAIP8抑制凋亡。同时,发现SMAD1抑制剂dorsomorphin(DM)与BTZ协同抑制耐药MM,为临床治疗提供了新策略。
3. 研究思路总结与详细解析
本研究以“SMAD1的临床表达特征→细胞功能验证→分子机制解析→体内治疗验证”为核心逻辑,围绕“SMAD1是否为MM耐药的关键调控因子及潜在治疗靶点”的科学问题,结合数据库分析、临床样本、细胞实验及动物模型,形成完整的研究闭环。
3.1 临床样本与数据库分析SMAD1的表达及预后关联
实验目的是明确SMAD1在MM患者中的表达特征及与临床预后的关系。方法上,作者利用GEO数据库(GSE6477、GSE77539、GSE31161)分析不同疾病阶段MM患者的SMAD1 mRNA表达,结合APEX临床试验的GSE9782数据集评估其与生存期的关联,并通过ROC曲线分析SMAD1的诊断效能。结果显示:SMAD1在复发MM中的表达显著高于初诊MM(GSE77539,p=0.011)和正常供者(GSE6477,p=0.032);SMAD1高表达患者的中位无进展生存期(PFS)为92周(低表达组121.5周,p=0.0168),中位总生存期(OS)为477天(低表达组674天,p=0.0142)(n=528);ROC曲线显示,SMAD1区分复发与初诊MM的曲线下面积(AUC)为0.7439,具备一定的诊断价值。

3.2 细胞实验验证SMAD1对MM增殖、迁移及耐药的调控
实验目的是验证SMAD1对MM细胞生物学行为的直接调控作用。方法上,选取耐药MM细胞系(8226R5、MM1.R、OPM2 vel/R),通过siRNA敲低SMAD1或DM抑制SMAD1,进行软琼脂克隆形成(增殖)、Transwell迁移(迁移)、MTT细胞活力(耐药性)实验。结果显示:敲低或抑制SMAD1后,耐药细胞的软琼脂克隆数减少(p<0.01),Transwell迁移能力下降(p<0.05);SMAD1敲低增强了细胞对BTZ的敏感性,细胞活力随BTZ浓度升高显著降低(p<0.05)。
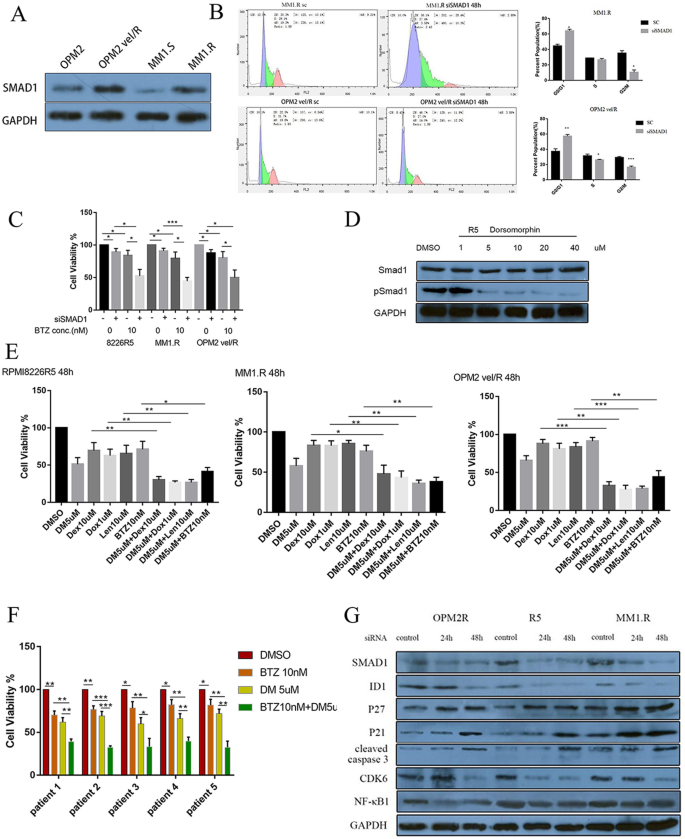
3.3 机制研究:SMAD1通过ID1/p21/p27轴促进MM增殖
实验目的是探索SMAD1促进增殖的分子机制。方法上,通过Western blot检测SMAD1敲低后ID1(inhibitor of DNA binding 1)及细胞周期蛋白p21、p27的表达变化。结果显示:SMAD1敲低后,ID1蛋白水平显著降低,而细胞周期抑制剂p21、p27的表达升高(图3h);进一步敲低ID1后,p21、p27表达上调,增殖受抑。表明SMAD1通过上调ID1抑制p21/p27,从而促进MM细胞增殖。
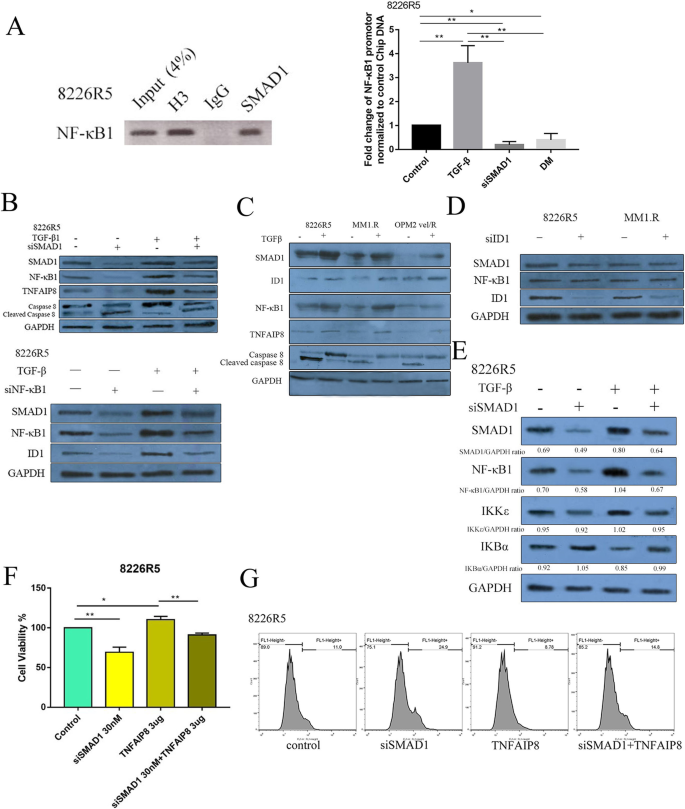
3.4 机制研究:SMAD1与NF-κB1的交互作用调控TNFAIP8抗凋亡
实验目的是揭示SMAD1抑制凋亡的分子机制。方法上,通过染色质免疫沉淀(ChIP)验证SMAD1与NF-κB1启动子的结合,利用Western blot、免疫荧光检测两者的相互调控,并通过rescue实验(转染TNFAIP8质粒)验证下游效应。结果显示:SMAD1直接结合NF-κB1启动子(图5a),两者表达呈正相关且相互调控(图4);SMAD1与NF-κB1共同促进TNFα诱导蛋白8(TNFAIP8)的表达,抑制caspase-8介导的凋亡(图5b、c);转染TNFAIP8可逆转SMAD1敲低引起的细胞活力下降和凋亡增加(图5f、g)。
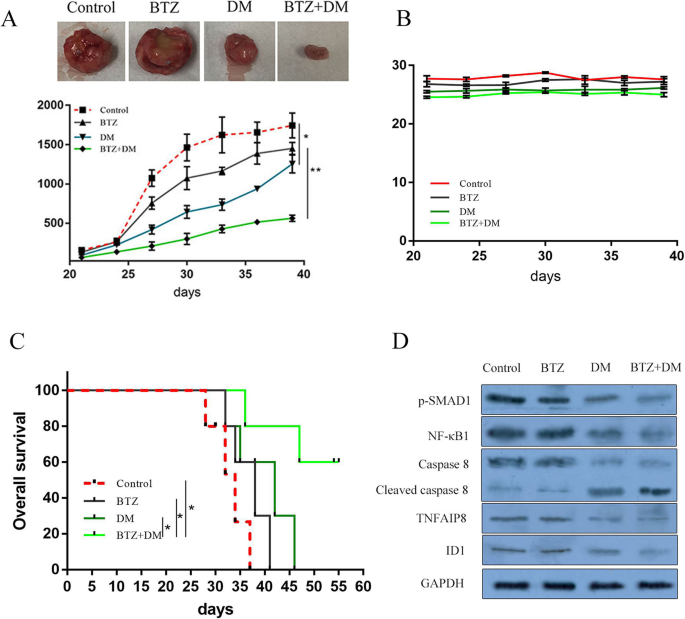
3.5 体内实验验证DM与BTZ的协同治疗效果
实验目的是验证SMAD1抑制剂的体内疗效。方法上,建立耐药MM细胞(OPM2 vel/R)的异种移植模型,给予DM和/或BTZ处理,监测肿瘤生长及生存期。结果显示:联合治疗组的肿瘤体积显著小于单药组(p<0.01),小鼠生存期显著延长(图6a、c);肿瘤组织中p-SMAD1、NF-κB1、TNFAIP8的表达降低,cleaved caspase-8升高(图6d),且DM对正常造血细胞无明显毒性(图S4C)。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究的核心生物标志物是SMAD1,作为MM耐药和预后的潜在生物标志物,其筛选与验证遵循“数据库挖掘→临床样本验证→功能实验确认”的逻辑链条:首先通过GEO数据库发现SMAD1在复发MM中高表达,接着利用APEX数据验证其与不良预后的关联,最后通过细胞/动物实验确认其调控耐药的功能。
研究过程详述
SMAD1的来源为MM患者的骨髓单个核细胞(BM-MNCs)、CD138+浆细胞及GEO数据库的基因表达数据;验证方法包括qRT-PCR(mRNA表达)、Western blot(蛋白表达)、ROC曲线(诊断效能)。特异性与敏感性数据显示:SMAD1区分复发与初诊MM的AUC为0.7439,区分初诊MM与正常供者的AUC为0.6706;预后关联数据显示,SMAD1高表达患者的中位PFS为92周(低表达组121.5周,p=0.0168),中位OS为477天(低表达组674天,p=0.0142)(n=528,高、低表达各264例)。
核心成果提炼
SMAD1是MM的不良预后生物标志物,其高表达与疾病进展、耐药及更短的生存期密切相关;作为功能型生物标志物,SMAD1通过调控ID1/p21/p27和NF-κB1/TNFAIP8轴促进MM耐药,是潜在的治疗靶点;临床转化潜力上,SMAD1抑制剂DM与BTZ联合可有效抑制耐药MM,为临床治疗提供了新策略。
综上,本研究首次系统解析了SMAD1在MM耐药中的作用及机制,为耐药MM的诊断与治疗提供了重要的生物标志物及靶点,具有显著的临床转化价值。