1. 领域背景与文献引入
文献英文标题:Dysregulation of JAK-STAT pathway in hematological malignancies and JAK inhibitors for clinical application;发表期刊:Biomark Res;影响因子:未公开;研究领域:血液系统恶性肿瘤中的信号通路调控与靶向治疗。
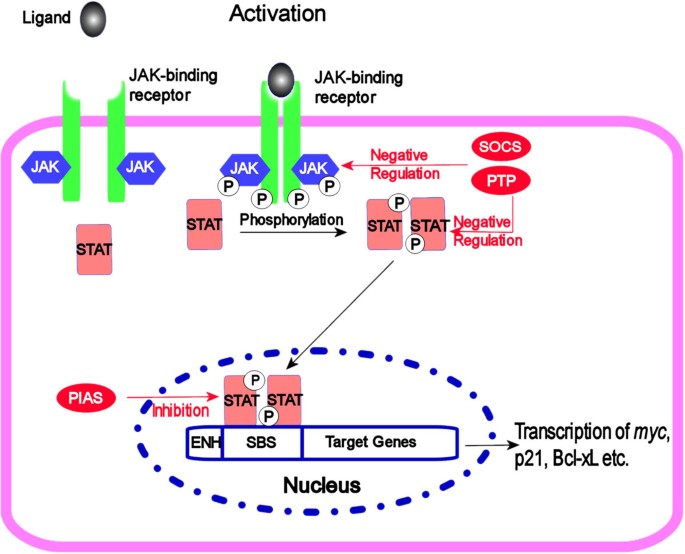
JAK-STAT(Janus激酶-信号转导与转录激活因子)通路是介导细胞因子、生长因子信号的关键 intracellular cascade,参与造血、免疫调节、胚胎发育等生理过程。通路核心组件包括4种JAK酪氨酸激酶(JAK1-3、Tyk2)和7种STAT转录因子(STAT1-6,含STAT5a/b),激活过程为“配体结合受体→JAK磷酸化→STAT二聚化核转位→靶基因转录”。负调控因子(如SHP1/2酪氨酸磷酸酶、PIAS家族蛋白、SOCS家族蛋白)通过去磷酸化、阻断DNA结合或促进泛素化降解维持通路平衡。
当前研究热点集中于:① 该通路在血液肿瘤中的失调机制(如 myeloproliferative neoplasms(MPN)中JAK2 V617F突变对STAT5的 constitutive激活);② JAK抑制剂的开发(如鲁索替尼成为首个获批用于骨髓纤维化的JAK1/2抑制剂)。未解决的核心问题包括:现有抑制剂选择性不足导致的不良反应(如贫血、血小板减少)、耐药性机制未明、不同突变亚型的精准治疗策略匮乏。
文献旨在系统梳理JAK-STAT通路在各类血液肿瘤中的失调模式,总结已获批及在研JAK抑制剂的临床数据,为基础研究与临床转化提供“机制-疾病-治疗”的完整参考,助力解决现有治疗瓶颈。
2. 文献综述解析
作者对现有研究的分类维度分为“通路调控机制”“疾病失调模式”“抑制剂临床进展”三部分,构建了“基础-临床”的逻辑链条。
现有研究的关键结论:① JAK2基因突变是MPN的核心驱动事件——JAK2 V617F突变在真性红细胞增多症(PV)中阳性率达80%,在原发性血小板增多症(ET)、原发性骨髓纤维化(PMF)中为30-50%,且等位基因 burden与表型相关(高 burden 对应PV,低 burden 对应ET);② 负调控因子异常(如SOCS-1双等位基因突变)会导致JAK2降解延迟、STAT5持续激活,与原发性纵隔B细胞淋巴瘤、霍奇金淋巴瘤发生相关;③ JAK抑制剂可缓解肿瘤负荷——鲁索替尼在骨髓纤维化患者中使41%患者脾脏缩小≥35%(安慰剂组为0%,P<0.001),且疗效与JAK2突变状态无关。
现有研究的技术优势在于结合“临床样本测序+细胞系功能验证+动物模型”(如JAK2 V617F转基因小鼠重现MPN表型),确保机制与临床的关联性;局限性则包括部分STAT激活的病理意义未明(如慢性淋巴细胞白血病中STAT1/3丝氨酸磷酸化是否直接致病)、抑制剂临床试验样本量小(如鲁索替尼早期试验仅纳入153例患者)。
文献的创新价值:① 全面覆盖MPN、淋巴瘤、白血病等多类血液肿瘤,弥补了现有综述对部分亚型(如急性淋巴细胞白血病)的关注不足;② 系统总结10余种JAK抑制剂的临床数据,分析选择性(如SAR302503为选择性JAK2抑制剂)与疗效的关联;③ 强调“突变类型-抑制剂选择”的精准理念,为不同患者群体的治疗优化提供依据。
3. 研究思路总结与详细解析
文献作为综述性研究,整体框架为“通路机制回顾→疾病失调分析→抑制剂临床总结”,核心科学问题是“JAK-STAT通路失调如何驱动血液肿瘤,及如何通过抑制剂靶向干预”。
3.1 JAK-STAT通路调控机制的系统回顾
实验目的:明确通路“激活-终止”的平衡机制,为疾病分析奠定基础。
方法细节:整合经典分子生物学研究(如co-IP验证JAK与受体结合、Western blot检测STAT磷酸化、荧光素酶报告基因分析转录活性)及负调控因子功能研究(如siRNA敲低SOCS-1观察JAK2降解)。
结果解读:通路正调控依赖“JAK-STAT”级联反应,负调控通过“去磷酸化(SHP1)、阻断DNA结合(PIAS3)、泛素化降解(SOCS-1)”实现。例如,SOCS-1通过SH2域结合JAK2,招募E3泛素连接酶促进其降解,终止信号。
产品关联:文献未提及具体实验产品,领域常规使用细胞因子(如重组人EPO)、磷酸化抗体(如抗p-JAK2、p-STAT5)、基因编辑工具(如siRNA)。
3.2 血液肿瘤中通路失调的多类型分析
实验目的:解析不同血液肿瘤中通路的异常模式,明确驱动突变与表型的关联。
方法细节:整合临床样本测序(如PCR检测JAK2 V617F、下一代测序检测SOCS-1突变)、细胞系功能实验(如JAK2 V617F转入Ba/F3细胞验证 cytokine independence)、动物模型研究(如JAK2 V617F转基因小鼠观察MPN表型)。
结果解读:① MPN中,JAK2 V617F通过解除JH2域对JH1激酶域的抑制,导致STAT5持续激活,促进红细胞过度增殖;JAK2 exon 12突变(如delV536)则通过改变JH2结构激活通路,多见于JAK2 V617F阴性PV患者。② 淋巴瘤中,霍奇金淋巴瘤的SEC31A-JAK2融合基因可 constitutive激活JAK2,原发性纵隔B细胞淋巴瘤的SOCS-1双等位基因突变会延迟JAK2降解,均导致STAT5持续激活。③ 白血病中,急性淋巴细胞白血病(ALL)的JAK2 R683突变、急性髓系白血病(AML)的STAT3激活(20-50%患者)与细胞增殖障碍相关。
产品关联:文献未提及具体实验产品,领域常规使用基因测序试剂盒(如Illumina MiSeq)、荧光原位杂交(FISH)探针(如JAK2探针)。
3.3 JAK抑制剂的临床进展梳理
实验目的:总结JAK抑制剂的疗效、安全性及耐药性。
方法细节:回顾I/II期剂量递增试验(评估最大耐受剂量)、III期随机对照试验(如COMFORT-I/II试验评估鲁索替尼对骨髓纤维化的疗效)。例如,鲁索替尼I/II期试验纳入153例骨髓纤维化患者,评估血小板减少的剂量限制性毒性;SAR302503 I/II期试验纳入59例MPN患者,分析JAK2 V617F等位基因 burden变化。
结果解读:① 鲁索替尼(非选择性JAK1/2抑制剂)在骨髓纤维化中显著缩小脾脏(41%患者缩小≥35%),但不良反应包括血小板减少(31%)、贫血(29%);② 选择性JAK2抑制剂(如SAR302503)可降低JAK2 V617F等位基因 burden(治疗6个月后,突变阳性患者burden显著下降,P=0.04),且对血小板影响较小;③ lestaurtinib(JAK2/FLT3抑制剂)在JAK2突变骨髓纤维化患者中脾脏反应率为27%,但血栓事件发生率较高(6/39例)。
产品关联:文献提及的关键产品包括鲁索替尼(Jakafi,Incyte)、SAR302503(TG101348)、lestaurtinib(CEP701)、CYT387、pacritinib(SB1518)等。
4. Biomarker研究及发现成果解析
文献涉及的Biomarker分为三类:① JAK家族基因突变(JAK2 V617F、exon 12突变、融合基因);② 负调控因子异常(SOCS-1突变);③ STAT激活(STAT3磷酸化、STAT5二聚化)。筛选逻辑遵循“临床样本发现→细胞系验证→动物模型确认→临床关联”流程。
Biomarker来源为血液肿瘤患者的临床样本(骨髓、肿瘤组织),验证方法包括:① 基因水平(PCR测序、FISH检测融合基因);② 蛋白水平(Western blot检测磷酸化、免疫组化检测核定位);③ 功能水平(细胞系 cytokine independence实验、动物模型致瘤性验证)。
特异性与敏感性数据:① JAK2 V617F在PV中的敏感性80%、特异性高(正常人群罕见);② JAK2 exon 12突变在JAK2 V617F阴性PV患者中敏感性约90%;③ SOCS-1双等位基因突变在原发性纵隔B细胞淋巴瘤中阳性率30%、特异性100%。
核心成果:① JAK2 V617F是MPN的预后Biomarker——等位基因 burden与表型相关(高 burden 对应PV),且高 burden 患者对SAR302503反应更好(P<0.01);② SOCS-1突变是霍奇金淋巴瘤的驱动事件,可预测JAK抑制剂疗效(突变患者更敏感);③ STAT3激活是AML的潜在预后Biomarker(20-50%患者存在激活,与不良预后相关)。
创新性在于首次系统总结了“突变Biomarker-抑制剂疗效”的关联,为精准治疗提供依据(如JAK2 V617F高 burden 患者优先选择选择性JAK2抑制剂)。