1. 领域背景与文献引入
文献英文标题:A functional role of Ephrin type-B receptor 6 (EPHB6) in T-cell acute lymphoblastic leukemia;发表期刊:Biomark Res;影响因子:未公开;研究领域:T细胞急性淋巴细胞白血病(T-cell acute lymphoblastic leukemia, T-ALL)发病机制与白血病起始细胞(leukemia initiating cells, LICs)靶向治疗。
T-ALL是一种侵袭性血液系统恶性肿瘤,以T细胞异常增殖为特征,尽管标准化疗显著提高了缓解率,但复发率仍高达15%~20%,且复发后患者5年生存率不足30%。其核心耐药机制在于肿瘤中存在少量具有自我更新能力的LICs,这类细胞对化疗不敏感且能驱动疾病复发。近年研究发现,Eph受体酪氨酸激酶(Eph receptor tyrosine kinases)家族在多种癌症干细胞中高表达,通过调控细胞增殖、迁移等信号通路参与肿瘤进展,但Eph家族成员EPHB6在T-ALL中的表达特征、与LICs的关联及临床意义尚未被系统阐明。已有研究仅提及EPHB6在儿童T-ALL中过表达,并可能影响肿瘤细胞对阿霉素的敏感性,但未深入探讨其在LICs维持及疾病进展中的功能。针对这一研究空白,本文旨在通过多层面实验明确EPHB6在T-ALL中的表达模式、与LICs的关系及临床预后价值,为T-ALL的靶向治疗提供新的分子靶点。
2. 文献综述解析
本文综述部分围绕“Eph受体家族与癌症干细胞的关联”核心逻辑展开,重点评述了Eph受体在肿瘤进展中的作用及EPHB6在T-ALL中的研究现状。现有研究表明,Eph受体家族(含16种受体)通过与ephrin配体结合,参与调控肿瘤细胞增殖、凋亡及干细胞维持,其中EphA2已被证实是胶质母细胞瘤干细胞的关键调控因子。针对T-ALL,前期研究发现EPHB6在儿童患者中过表达,且其表达水平与阿霉素敏感性相关,但EPHB6是否参与LICs的维持、是否可作为临床预后标志物等关键问题尚未解决。
本文的创新价值在于:首次系统整合公共数据集、细胞系、患者来源异种移植(patient-derived xenografts, PDX)模型及临床样本单细胞测序数据,明确了EPHB6作为T-ALL LICs特异性标志物的功能——不仅填补了EPHB6在T-ALL LICs研究中的空白,还将其表达与临床预后直接关联,为靶向LICs的精准治疗提供了理论依据。
3. 研究思路总结与详细解析
本研究以“明确EPHB6在T-ALL中的表达特征→验证其对LICs的调控作用→解析分子机制→关联临床预后”为核心逻辑,采用“公共数据集分析→细胞系功能验证→PDX模型体内实验→单细胞RNA-seq临床样本分析”的闭环技术路线,逐步阐明EPHB6的功能及临床意义。
3.1 公共RNA-seq数据集的EPHB6表达分析
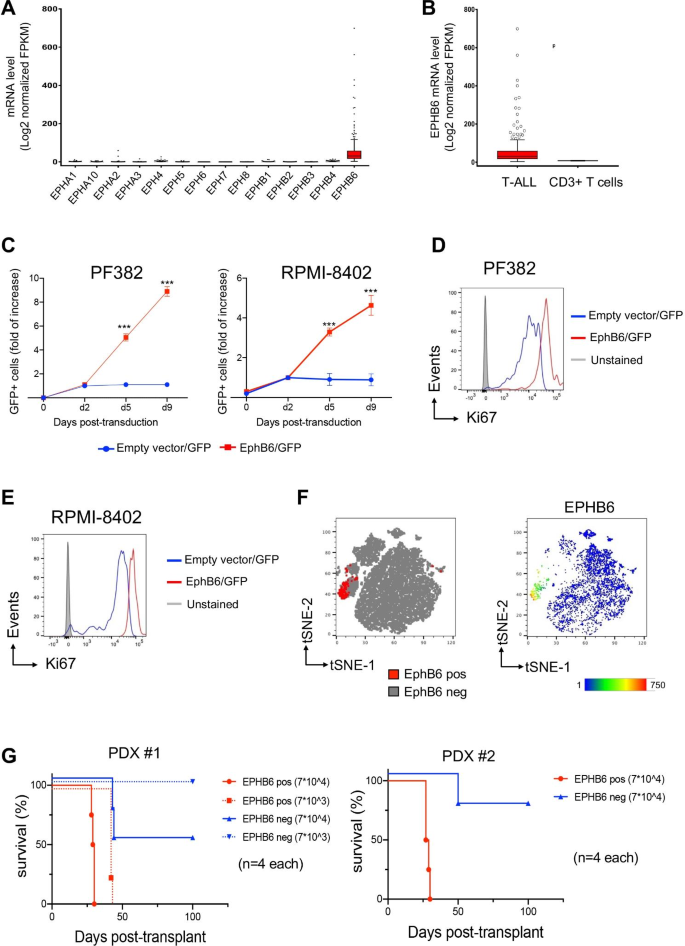
实验目的是明确EPHB6在T-ALL患者中的表达特征及与其他Eph家族成员的差异。方法为:分析COG TARGET公共数据集(264例T-ALL患者)的RNA-seq数据,比较Eph家族成员的表达水平;同时利用Human Protein Atlas数据库(20例健康供者CD3+ T细胞),对比T-ALL患者与健康人群的EPHB6表达差异。结果显示:EPHB6是Eph家族中唯一在T-ALL患者中显著过表达的成员(与其他Eph受体相比,P<0.0001,n=264);且T-ALL患者的EPHB6表达水平显著高于健康供者的CD3+ T细胞(P<0.0001,n=20)。
文献未提及具体实验产品,领域常规使用RNA-seq数据分析工具(如DESeq2、EdgeR)及公共数据库(如GEO、TCGA)。
3.2 细胞系中EPHB6的增殖功能验证
实验目的是验证EPHB6对T-ALL细胞增殖的调控作用。方法为:构建EPHB6过表达慢病毒载体(EPHB6组)及空载体(EV组),转导T-ALL细胞系PF382和RPMI-8402;通过流式细胞术追踪GFP+活细胞比例(DAPI排除法区分活细胞),并检测细胞增殖标志物Ki67的表达水平。结果显示:与EV组相比,EPHB6过表达显著促进细胞扩张(如PF382细胞在培养第7天的GFP+活细胞比例较EV组高2.1倍,P<0.001,n=3);同时,EPHB6组细胞的Ki67表达水平显著升高(PF382细胞Ki67阳性率为68.2% vs EV组的35.1%,P<0.001,n=3)。
文献未提及具体实验产品,领域常规使用慢病毒载体系统(如pLVX-Puro)、流式细胞仪(如BD FACSCanto II)及Ki67单克隆抗体(如Abcam ab15580)。
3.3 PDX模型中EPHB6+细胞的LIC活性验证
实验目的是明确EPHB6+细胞的白血病起始能力。方法为:将T-ALL患者活检样本注射到免疫缺陷NSG小鼠中,构建PDX模型(M71和H3255两个独立克隆);通过多参数流式细胞术分选EPHB6+和EPHB6-细胞,将不同剂量的分选细胞移植到NSG小鼠中,观察小鼠生存期。结果显示:EPHB6+细胞移植的小鼠生存期显著短于EPHB6-细胞组(如M71克隆中,注射1×10^4 EPHB6+细胞的小鼠中位生存期为35天,而EPHB6-细胞组为62天,P<0.01,n=4);且EPHB6+细胞的LIC频率显著高于EPHB6-细胞(M71克隆中EPHB6+细胞的LIC频率为1/125,而EPHB6-细胞为1/1000)。
实验所用关键产品:NSG免疫缺陷小鼠(Jackson Laboratory);流式细胞术抗体信息见补充表S1(文献未提及具体品牌)。
3.4 转录组与单细胞RNA-seq的临床意义分析
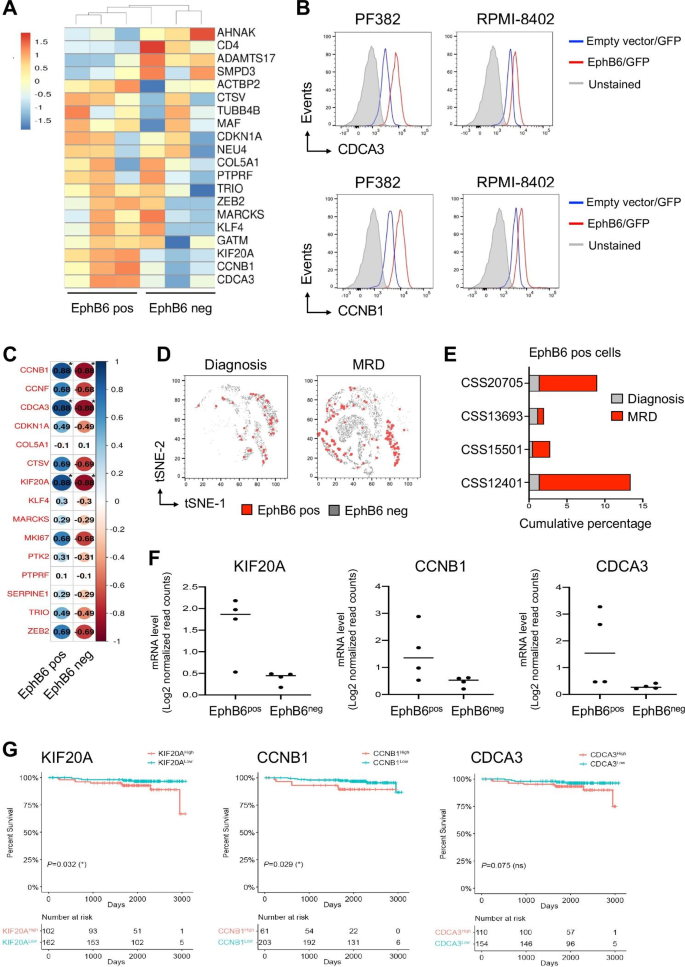
实验目的是解析EPHB6+细胞的分子特征及临床治疗后的富集情况。方法为:对PDX模型的EPHB6+/-细胞进行RNA-seq,筛选差异表达基因;对4例T-ALL患者诊断时(d0)和治疗30天( minimal residual disease, MRD)的样本进行单细胞RNA-seq,分析EPHB6+细胞的比例变化及基因表达特征;利用COG TARGET数据集分析差异基因与患者生存的关联。结果显示:EPHB6+细胞高表达细胞周期相关基因(如CCNB1、CDCA3、KIF20A);且在MRD样本中,EPHB6+细胞比例显著高于诊断时(如CSS12401样本中,d0时EPHB6+细胞比例为12.3%,MRD时为35.6%,P<0.001,n=4);高表达KIF20A和CCNB1的患者生存期显著缩短(KIF20A高表达组中位生存期为36个月 vs 低表达组的58个月,P=0.032;CCNB1高表达组中位生存期为38个月 vs 低表达组的60个月,P=0.029,n=264)。
文献未提及具体实验产品,领域常规使用单细胞测序平台(如10x Genomics Chromium)及生存分析工具(如R软件survival包)。
4. Biomarker研究及发现成果解析
Biomarker定位与筛选逻辑
本文鉴定的Biomarker为EPHB6,其作为T-ALL LICs的特异性标志物,筛选与验证逻辑为“公共数据集初筛→细胞系功能验证→PDX模型体内验证→临床样本单细胞分析”:首先通过公共RNA-seq数据集明确EPHB6在T-ALL中的过表达特征;接着利用细胞系验证其对增殖的调控作用;再通过PDX模型证实其与LICs的关联;最后通过临床样本单细胞测序明确其在MRD中的富集及预后价值。
研究过程详述
EPHB6的来源包括:T-ALL患者肿瘤样本(公共数据集)、PDX模型细胞及临床治疗前后样本(诊断时和MRD)。验证方法涵盖:① 公共数据集的RNA-seq分析(检测表达水平);② 细胞系与PDX模型的流式细胞术(检测蛋白表达);③ 单细胞RNA-seq(分析临床样本中的细胞比例及基因表达)。特异性与敏感性方面,EPHB6+细胞在PDX模型中显示出更高的LIC活性(生存期更短、LIC频率更高),且在MRD中显著富集(各样本MRD中EPHB6+细胞比例较诊断时高2~3倍,文献未明确具体AUC值)。
核心成果提炼
EPHB6作为T-ALL LICs的新型Biomarker,其核心功能与临床价值包括:① 功能关联:EPHB6+细胞是T-ALL的LICs亚群,通过高表达细胞周期基因(如CCNB1、KIF20A)维持自我更新和增殖能力;② 临床意义:EPHB6+细胞在MRD中富集,提示其与化疗耐药相关;③ 预后价值:高表达EPHB6关联的基因(KIF20A、CCNB1)与患者低生存显著相关(KIF20A:HR=0.70,95%CI 0.54-0.91,P<0.01;CCNB1:HR=0.53,95%CI 0.39-0.71,P<0.001)。
本研究首次明确EPHB6作为T-ALL LICs标志物的作用,为T-ALL的靶向治疗提供了新的分子靶点——针对EPHB6+细胞的治疗策略或可提高化疗疗效,降低复发率。