1. 领域背景与文献引入
文献英文标题:Inappropriate costimulation and aberrant DNA methylation as therapeutic targets in angioimmunoblastic T-cell lymphoma;发表期刊:Biomarker Research;影响因子:未公开;研究领域:血管免疫母细胞性T细胞淋巴瘤(AITL)的分子机制与治疗靶点。
外周T细胞淋巴瘤(PTCL)是一组异质性非霍奇金淋巴瘤,以侵袭性临床进程和 poor 预后为特征。血管免疫母细胞性T细胞淋巴瘤(AITL)是PTCL最常见的亚型之一,占所有病例的15–20%。尽管对AITL的突变谱理解有所进展,但过去二十年患者预后并未改善(5年总生存率25–41%),且一线和二线治疗均无共识。近年来提出的AITL多步骤致癌模型为其发病机制提供了理论框架:早期造血干细胞(HSC)或成熟T细胞获得表观遗传修饰基因(如TET2、IDH2、DNMT3A)突变,导致克隆性造血或T细胞分化异常;晚期获得协同突变(如RHOA、CD28),驱动恶性转化。现有研究分别聚焦于异常DNA甲基化(表观遗传修饰基因缺陷)和不适当共刺激(T细胞受体或共刺激通路激活)的分子机制,但尚未探索针对这两个通路的联合治疗策略。本文作为评论文章,旨在总结这两个关键通路的研究进展,提出联合靶向治疗的假设,为AITL治疗提供新方向。
2. 文献综述解析
作者将现有AITL研究分为“异常DNA甲基化驱动早期事件”和“不适当共刺激介导晚期转化”两大方向,通过对比现有研究的局限性,凸显本文的创新价值。
核心评述逻辑与现有研究总结
作者以“多步骤致癌模型”为核心,将现有研究拆解为两个互补的分子通路:
1. 异常DNA甲基化的早期事件:测序研究显示,TET2(30–80%)、IDH2(20–45%)、DNMT3A(10–30%)在AITL中高频突变。TET2是DNA去甲基化酶,突变导致HSC自我更新增强、分化异常,并通过甲基化BCL6基因内含子1(沉默区)诱导BCL6过表达(T滤泡辅助细胞(Tₚₕ)的谱系定义转录因子);IDH2 R172残基突变产生致癌代谢物(R)-2-羟基戊二酸(2HG),抑制TET2功能,减少DNA羟甲基化;DNMT3A是从头甲基转移酶,突变导致HSC分化障碍,与TET2突变协同促进淋巴恶性转化(小鼠模型显示联合突变可诱导AITL样疾病)。这些研究明确了表观遗传异常在AITL早期发病中的作用,但未探索其与晚期协同突变的相互作用。
2. 不适当共刺激的晚期转化:CD28突变(T195P、D124V)在5–11%的AITL中出现,减少与PI3K p85/p110异二聚体的结合,但增加与Grb2/GRAP2的结合,增强下游ERK和Akt磷酸化,促进细胞增殖,且突变患者预后更差;CTLA4-CD28融合基因在58%的亚洲队列AITL中存在,融合蛋白以CTLA-4的高亲和力结合配体,同时介导CD28的激活信号,增加Akt和ERK磷酸化;RHOA G17V突变在50–70%的AITL中出现,通过增强诱导性T细胞共刺激因子(ICOS)信号诱导Tₚₕ表型,且小鼠模型显示突变RHOA与TET2缺陷协同导致AITL,肿瘤细胞依赖ICOS信号生存。这些研究揭示了共刺激通路异常在晚期转化中的作用,但缺乏与表观遗传异常的联合靶向研究。
本文创新价值
现有研究多聚焦单一通路的机制解析,未关注“异常DNA甲基化-不适当共刺激”的协同作用:作者通过整合研究发现,异常DNA甲基化可下调T细胞受体信号负调节剂(如蛋白酪氨酸磷酸酶非受体型7(PTPN7)、SIT1、二酰甘油激酶α(DGKA)),增强不适当共刺激的下游信号(如Ras/Raf/MAPK/ERK),形成正反馈环路。而现有研究未针对这一协同机制设计治疗方案。本文的创新点在于首次提出联合靶向异常DNA甲基化(去甲基化药物)和不适当共刺激(CTLA-4阻滞剂、PI3K/mTOR抑制剂)的治疗策略,为AITL治疗提供新的理论框架。
3. 研究思路总结与详细解析
作为评论文章,作者未开展原创实验,而是通过“机制回顾-靶点挖掘-治疗假设”的逻辑框架,系统整合现有研究,具体思路如下:
3.1 AITL多步骤致癌模型的分子基础回顾
作者首先梳理多步骤致癌模型的核心观点:早期表观遗传修饰基因(TET2、IDH2、DNMT3A)突变导致细胞分化异常,晚期协同突变(RHOA、CD28等)驱动恶性转化。并引用关键实验验证模型的合理性:例如,TET2缺陷小鼠的HSC自我更新增强,联合RHOA G17V突变可诱导AITL(小鼠模型);RHOA G17V突变的CD4⁺ T细胞通过ICOS信号诱导Tₚₕ表型,抗ICOS抗体可逆转该表型(功能实验)。
3.2 异常DNA甲基化的机制解析
作者逐一解析表观遗传修饰基因的功能及突变影响:
- TET2突变:通过甲基化BCL6内含子1(沉默区)抑制其转录抑制功能,导致BCL6过表达,驱动Tₚₕ分化(Nishizawa等,2016年研究);
- IDH2 R172突变:产生2HG,抑制TET2和其他2-氧戊二酸(2OG)依赖酶,减少DNA羟甲基化,影响T细胞发育(Lemonnier等,2016年研究);
- DNMT3A突变:与TET2突变协同,通过异常甲基化调控干细胞自我更新基因(如HOX家族),增加恶性转化风险(Scourzic等,2016年研究)。
这些机制解析基于测序数据(如TET2突变在30–80%的AITL中出现)、功能实验(如TET2敲低细胞的BCL6表达检测)和临床关联(如TET2突变患者对去甲基化药物反应好)。
3.3 不适当共刺激的机制解析
作者聚焦共刺激通路的异常激活:
- CD28突变/融合:CD28 T195P突变通过Grb2/GRAP2增强ERK和Akt磷酸化(Lee等,2015年研究);CTLA4-CD28融合蛋白结合CD80/CD86后,激活PI3K/Akt/mTOR通路(Yoo等,2016年研究);
- RHOA突变:RHOA G17V通过增强ICOS信号稳定BCL6,诱导Tₚₕ表型,且小鼠模型显示突变细胞依赖ICOS信号生存(Cortes等,2016年研究)。
这些机制基于功能实验(如突变CD28转染细胞的增殖检测)和临床数据(如CD28突变患者生存期缩短)。
3.4 靶向治疗策略的提出
基于上述机制,作者提出两类治疗策略:
1. 靶向异常DNA甲基化:使用去甲基化药物(阿扎胞苷、地西他滨)抑制DNA甲基转移酶(DNMTs),逆转异常甲基化模式。例如,12例复发/难治(R/R)AITL患者使用阿扎胞苷治疗,总反应率75%,完全缓解率42%(其中8例TET2突变患者均有反应,Delarue等,2016年研究);
2. 靶向不适当共刺激:①阻断配体-共受体相互作用:CTLA-4阻滞剂(伊匹木单抗、阿巴西普)或ICOS阻滞剂;②抑制下游信号:PI3Kδ抑制剂(艾代拉里斯)、mTOR抑制剂(依维莫司、替西罗莫司)。例如,依维莫司联合CHOP治疗新诊断PTCL,3例AITL患者均完全缓解(Kim等,2016年研究)。
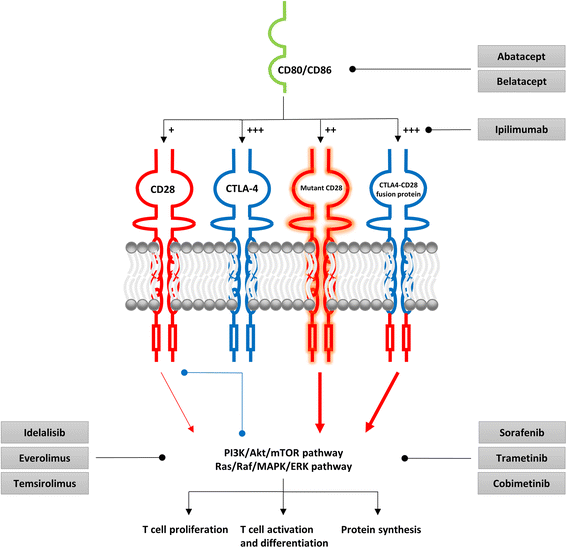
3.5 联合治疗的理论依据
作者提出,异常DNA甲基化通过下调T细胞受体信号负调节剂(如PTPN7)增强不适当共刺激的下游信号,而联合治疗可通过以下机制协同作用:
- 去甲基化药物恢复负调节剂的表达(如PTPN7),削弱共刺激信号的下游传导;
- 共刺激抑制剂直接阻断异常激活的通路(如CTLA-4阻滞剂抑制CD28/CTLA4-CD28融合蛋白的配体结合);
- 两者联合可打破正反馈 loop,更有效地抑制肿瘤细胞增殖。
例如,IDH2 R172突变患者中,异常甲基化导致PTPN7(MAPK特异性磷酸酶)下调,增强Ras/Raf/MAPK/ERK信号,联合使用阿扎胞苷(恢复PTPN7表达)和MEK抑制剂(抑制ERK)可能更有效。
4. Biomarker研究及发现成果解析
4.1 潜在Biomarker的筛选与验证
文中涉及的潜在Biomarker均为AITL中高频突变的基因,筛选与验证逻辑如下:
- 表观遗传修饰基因:TET2、IDH2(R172)、DNMT3A,通过大规模测序研究(如全外显子测序)发现高频突变(TET2 30–80%、IDH2 20–45%、DNMT3A 10–30%),功能实验验证突变对DNA甲基化和基因表达的影响(如TET2突变导致BCL6过表达),临床关联分析验证对去甲基化药物的反应(如TET2突变患者对阿扎胞苷反应好);
- 协同突变基因:RHOA(G17V)、CD28(T195P/D124V)、CTLA4-CD28融合基因,通过测序研究发现高频突变/融合(RHOA 50–70%、CD28 5–11%、融合基因58%亚洲队列),功能实验验证突变对下游信号的激活(如RHOA突变增强ICOS信号),临床关联分析验证对预后的影响(如CD28突变患者生存期缩短)。
4.2 Biomarker的功能与临床意义
这些Biomarker不仅是AITL的分子诊断标志,还具有功能关联性和治疗指导价值:
1. TET2突变:作为去甲基化药物的疗效预测Biomarker,12例R/R患者中8例TET2突变者均对阿扎胞苷有反应(无具体P值,但临床反应显著);
2. IDH2 R172突变:作为2HG产生和TET2抑制的标志,可预测对TET2激活剂或2HG抑制剂的反应;
3. RHOA G17V突变:作为ICOS信号增强的标志,可预测对ICOS阻滞剂的反应;
4. CD28突变/CTLA4-CD28融合基因:作为共刺激信号激活的标志,可预测对CTLA-4阻滞剂或PI3K/mTOR抑制剂的反应。
作者强调,这些Biomarker的临床验证仍需大规模前瞻性研究,但现有数据支持其作为个性化治疗的潜在靶点,例如:TET2突变+RHOA突变的患者可联合使用阿扎胞苷和ICOS阻滞剂,CD28融合基因患者可联合使用CTLA-4阻滞剂和依维莫司。
