1. 领域背景与文献引入
文献英文标题:DeepMPTB: a vaginal microbiome-based deep neural network as artificial intelligence strategy for efficient preterm birth prediction;发表期刊:Biomarker Research;影响因子:未公开;研究领域:早产预测与阴道微生物组。
早产(Preterm Birth, PTB)是全球新生儿死亡的主要原因,也是5岁以下儿童死亡的第二大诱因,早产儿常面临神经损伤、呼吸及胃肠道疾病等长期健康风险。现有早产诊断方法主要依赖孕妇产科史采集和孕早中期经阴道超声测量宫颈长度,但这些方法受医师经验差异影响大,且流程耗时、准确性有限。近年研究提示,阴道微生物组通过维持阴道酸性环境、抑制致病菌生长、调节炎症反应(如跨 kingdom 信号传导)等机制,可能参与早产的病理生理过程。然而,尽管已有多项宏基因组学研究分析阴道微生物组与早产的关联,但尚无明确、统一的微生物特征可用于临床早产预测,且不同研究队列的微生物组谱异质性大,结果难以重复。针对现有诊断方法的局限性及阴道微生物组研究的空白,本研究旨在整合多队列阴道宏基因组数据与临床数据,开发基于深度神经网络(Deep Neural Network, DNN)的早产预测模型DeepMPTB,以提升早产预测的效率与准确性。
2. 文献综述解析
文献综述围绕“现有早产诊断方法的局限性”与“阴道微生物组-早产关联的研究现状”两大核心展开评述。现有研究的关键结论包括:早产是全球新生儿健康的重大威胁,现有诊断方法依赖主观经验且准确性不足;阴道微生物组的平衡状态与妊娠结局密切相关,其功能紊乱可能诱导炎症反应,进而触发早产。技术方法上,部分研究采用宏基因组学技术解析阴道微生物组组成,但局限在于未形成明确的微生物预测特征,且不同队列的微生物组谱异质性高,导致结果难以推广。
与现有研究相比,本研究的创新点体现在三方面:一是整合5个独立研究队列的1290份阴道样本数据(覆盖足月与早产群体),显著扩大了样本量与队列多样性;二是采用深度神经网络模型替代传统机器学习算法,更擅长捕捉微生物组与临床数据间的复杂非线性关系;三是突破“寻找特定微生物物种”的传统思路,强调阴道微生物组的整体多样性而非单一物种的作用,并纳入临床元数据(如年龄、种族、采样 trimester)以提升模型性能。
3. 研究思路总结与详细解析
本研究的整体框架为“数据收集→微生物组分析→模型训练→性能验证→结论”,核心目标是构建“阴道微生物组+临床数据”的深度神经网络模型,解决“如何高效利用阴道微生物组数据预测早产”的科学问题,技术路线形成“数据输入→模型优化→验证输出”的闭环。
3.1 数据收集与微生物组分析
实验目的是获取高质量的阴道微生物组与临床数据,为模型训练提供基础。方法细节上,研究收集5个独立队列的1290份阴道样本(来自561名孕妇,其中1029份为足月分娩者,261份为早产者),通过RiboTaxa pipeline结合SILVA SSU 138.1 NR99数据库分析宏基因组fastq文件,生成物种水平的微生物组谱(涵盖细菌与真核生物);同时收集每个样本的临床元数据(年龄、种族、采样所处的妊娠 trimester)。结果解读显示,不同队列内的阴道微生物组谱差异显著(Welch’s t-test,p<0.05),体现了阴道微生物组的个体异质性,但足月与早产组的α多样性(微生物群落内部多样性)无显著差异;所有样本的微生物组谱与临床数据被整合为统一训练集,用于后续模型构建。
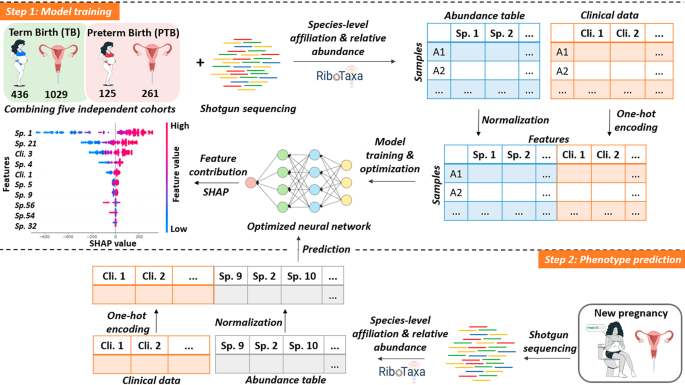
实验所用关键工具:RiboTaxa生物信息学软件、SILVA SSU 138.1 NR99数据库;领域常规使用宏基因组测序试剂盒(如Illumina TruSeq)进行样本测序。
3.2 深度神经网络模型训练与优化
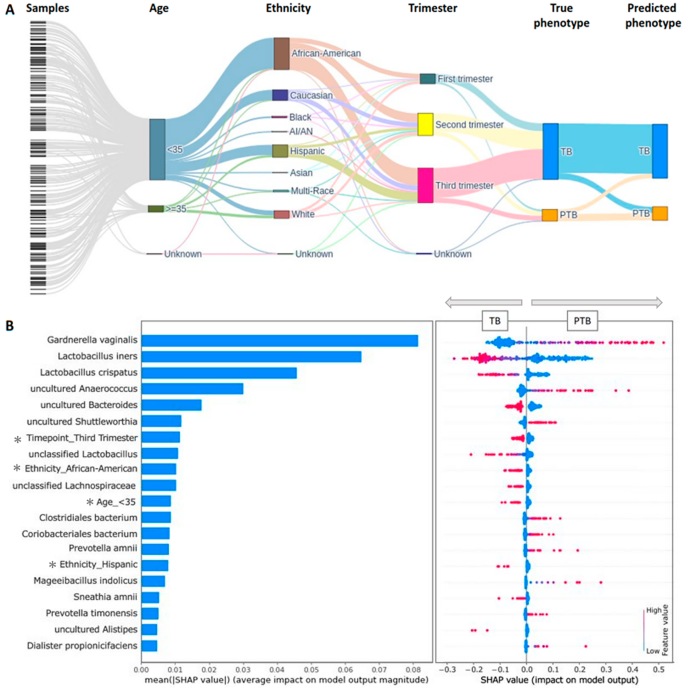
实验目的是构建并优化用于早产预测的深度神经网络模型。方法细节上,以“归一化的物种丰度数据+向量化的临床元数据”为输入,训练深度神经网络;通过超参数优化确定最终模型结构:包含3个隐藏层,第一层416个神经元,后续每层神经元数为前一层的一半,共234786个可训练参数;使用SHAP(SHapley Additive exPlanations)工具分析特征重要性,解释模型的预测逻辑。结果解读显示,优化后的模型在20%测试集(239份样本)上的准确性为84.10%,受试者工作特征曲线下面积(AUROC)为0.875±0.11;SHAP分析提取了20个关键预测特征,涵盖低丰度微生物物种(如某些罕见细菌)与临床元数据(如年龄、采样 trimester),提示低丰度物种与临床信息均对早产预测有重要贡献。
实验所用关键工具:SHAP解释器;领域常规使用TensorFlow或PyTorch框架进行深度神经网络训练。
3.3 模型性能基准测试与验证
实验目的是验证DeepMPTB的性能优于现有机器学习算法,并测试不同条件下的模型稳定性。方法细节上,将DeepMPTB与7种常用机器学习算法(决策树、K最近邻、随机森林、朴素贝叶斯、极端梯度提升、逻辑回归、支持向量机)进行基准测试;分别用孕第一、二、三 trimester的样本训练模型,比较预测准确性;使用RiboTaxa、DeepMicrobes、MetaPhlAn3三种宏基因组分类器处理数据,训练模型并比较性能;测试“仅用微生物组数据(无临床元数据)”的模型性能;最后用外部队列(694份样本,430份足月、264份早产)验证模型通用性。结果解读显示,DeepMPTB的受试者工作特征曲线下面积(0.877±0.11)显著优于其他算法(ANOVA test,p<0.05);第三 trimester样本训练的模型准确性最高(88%),第一、二 trimester分别为71%和83%;RiboTaxa处理的微生物组数据显著提升模型性能(受试者工作特征曲线下面积0.898±0.09,ANOVA test,p<0.05),优于DeepMicrobes(0.838±0.14)与MetaPhlAn3(0.795±0.08);仅用微生物组数据的模型性能下降(受试者工作特征曲线下面积0.831±0.12,Mann-Whitney U test,p>0.05);外部队列验证中,模型正确识别80%的足月样本与66%的早产样本。
实验所用关键工具:ANOVA、Mann-Whitney U test统计方法;领域常规使用Python的scikit-learn库实现传统机器学习算法。
4. Biomarker研究及发现成果解析
本研究的Biomarker为“阴道微生物组物种丰度特征(含低丰度物种)与临床元数据(年龄、种族、采样 trimester)的整合特征”,筛选逻辑是通过SHAP分析从深度神经网络中提取关键预测特征,验证逻辑则是通过“不同条件下的模型性能测试+外部队列验证”。
Biomarker的来源为5个队列的阴道宏基因组样本(物种丰度数据)与临床记录;验证方法包括SHAP特征重要性分析、模型基准测试(与其他算法对比)、不同孕期样本的性能测试、外部队列验证;特异性与敏感性数据方面,模型在测试集上的准确性为84.10%,受试者工作特征曲线下面积为0.875±0.11;外部队列中,足月样本识别率(特异性相关)为80%,早产样本识别率(敏感性)为66%。
核心成果方面,整合特征作为Biomarker的预测性能显著优于现有机器学习模型(ANOVA test,p<0.05);创新性在于突破“寻找特定微生物物种”的传统思路,强调阴道微生物组的整体多样性与临床数据的结合,通过深度神经网络捕捉复杂的微生物-临床数据相互作用;统计学结果显示,RiboTaxa处理的微生物组数据显著提升模型性能(ANOVA test,p<0.05),临床数据的加入虽未达统计学显著(Mann-Whitney U test,p>0.05),但仍能提升预测性能(受试者工作特征曲线下面积从0.831±0.12升至0.875±0.11)。此外,研究发现第三 trimester的样本更利于模型预测,提示采样时间可能影响Biomarker的有效性。
综上,本研究的Biomarker为早产预测提供了新的思路——“微生物组整体特征+临床数据”的整合策略,而非单一物种,这为后续临床转化提供了更稳健的基础。