1. 领域背景与文献引入
文献英文标题:FAM83A is a potential biomarker for breast cancer initiation;发表期刊:Biomarker Research;影响因子:未公开;研究领域:乳腺癌生物标志物与肿瘤起始机制。
乳腺癌是全球女性发病率最高的恶性肿瘤,早期诊断与干预是降低死亡率的核心策略,但目前缺乏能精准预测肿瘤起始的特异性生物标志物。家族序列相似性83成员A(FAM83A)是近年发现的致癌基因,在肺癌、胰腺癌、乳腺癌等多种肿瘤中过表达,可促进细胞增殖、侵袭及酪氨酸激酶抑制剂(TKI)耐药。作者前期研究发现,高风险乳腺癌女性的正常乳腺组织中FAM83A已呈过表达,提示其可能参与肿瘤起始过程。然而,FAM83A在乳腺癌早期阶段的功能及分子机制尚未明确,本文旨在验证“FAM83A是乳腺癌起始关键因子”的假设,探索其作为早期生物标志物的潜力。
2. 文献综述解析
作者通过三类研究总结现有认知:① FAM83A的泛癌致癌作用:在乳腺癌中,FAM83A过表达与HER2阳性、TKI耐药相关,可激活PI3K/AKT、MAPK等促癌通路;② FAM83A的临床关联:肿瘤组织中高表达与不良预后相关,但缺乏正常组织中的表达分析;③ 机制研究局限:现有研究多基于肿瘤细胞系,未探索其在正常乳腺上皮细胞中的功能。
现有研究的核心不足是未聚焦乳腺癌起始阶段——即从正常上皮细胞向癌细胞转化的早期过程。本文的创新点在于:1)利用大样本组织芯片(TMA)分析正常与肿瘤组织中FAM83A的表达及相关性;2)在原代与永生化乳腺上皮细胞中验证FAM83A的功能;3)鉴定不同细胞状态下FAM83A的相互作用蛋白,揭示机制差异。
3. 研究思路总结与详细解析
本文采用“临床组织分析→细胞功能验证→分子机制解析”的闭环思路,分6个关键实验环节展开:
3.1 组织芯片免疫组化分析
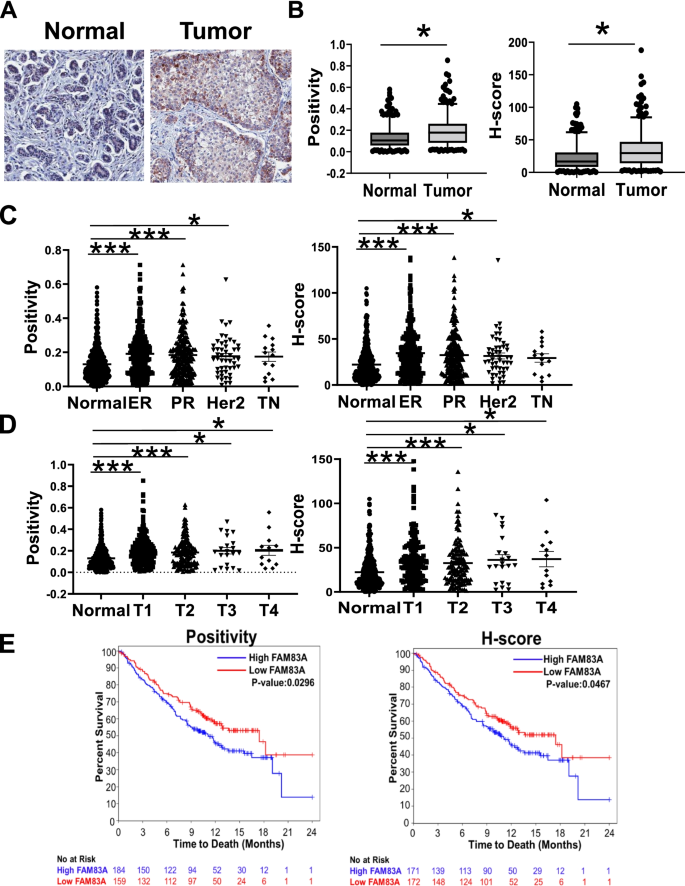
实验目的:评估FAM83A与EGFR在正常及肿瘤乳腺组织中的表达及相关性。
方法细节:使用正常乳腺TMA(N=411,来自Susan G. Komen组织库)和肿瘤TMA(N=349,来自IUSCCC组织库),通过免疫组化(IHC)检测FAM83A与EGFR蛋白表达。FAM83A抗体为Protein Tech的20618–1-AP(1:100),EGFR抗体为Agilent的K149489(1:400);采用Dako Link48系统进行染色,Aperio Scanscope CS成像并通过Positive Pixel Count算法计算H-score(半定量评分)。
结果解读:肿瘤组织中FAM83A的H-score较正常组织高1.5倍(n=349 vs 411,p<0.0001);且FAM83A与EGFR表达呈正相关——正常组织中皮尔逊相关系数r=0.4(p<0.0001),肿瘤组织中r=0.28(p<0.0001)。
产品关联:实验所用关键产品:Protein Tech的FAM83A抗体(货号20618–1-AP)、Agilent的EGFR抗体(货号K149489)、Dako Link48染色系统、Aperio成像分析软件。
3.2 高风险女性乳腺组织中FAM83A表达验证
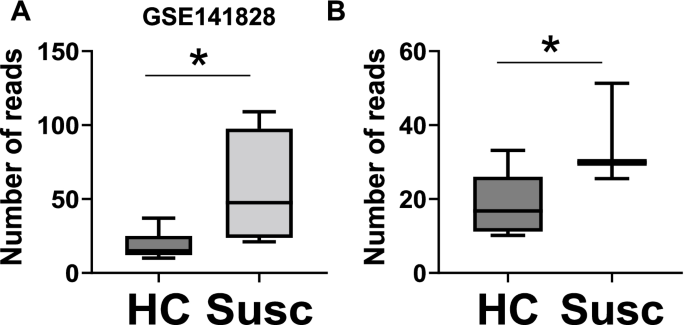
实验目的:确认FAM83A在乳腺癌起始阶段的过表达。
方法细节:分析GSE141828数据集(含4例“易感正常”组织——后续确诊乳腺癌的女性正常乳腺组织,8例健康对照),及独立队列(3例易感正常组织、8例健康对照)的转录组数据,评估FAM83A mRNA水平。
结果解读:易感正常组织中FAM83A表达较健康对照高3.9倍(n=4 vs 8,p=0.02);独立队列中高1.9倍(n=3 vs 8,p=0.03),提示FAM83A过表达发生在肿瘤确诊前的“起始阶段”。
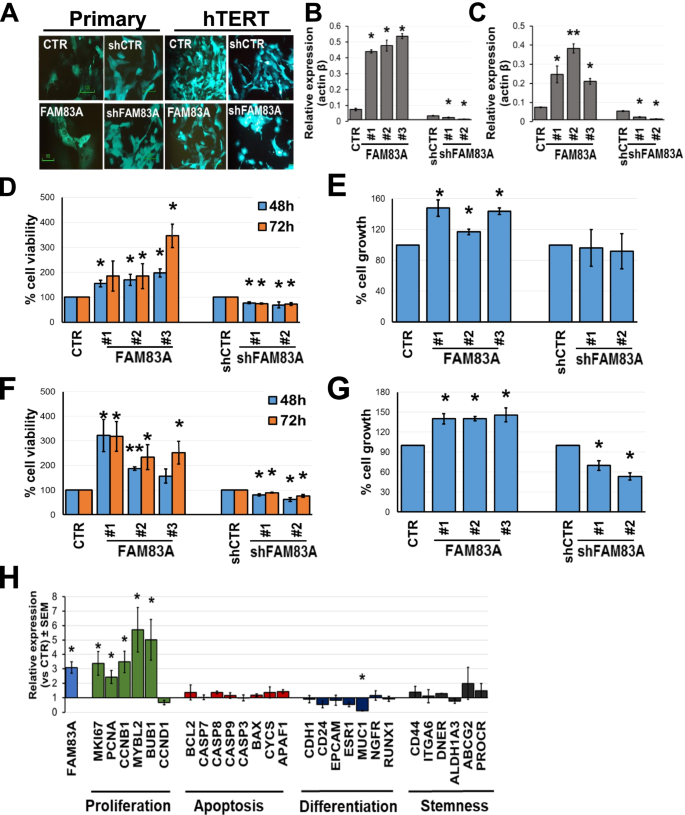
3.3 细胞模型构建与FAM83A表达调控
实验目的:建立可调控FAM83A表达的乳腺上皮细胞模型。
方法细节:从平均风险女性乳腺组织中分离原代上皮细胞,通过Amsbio的hTERT慢病毒(货号LVP1130)感染建立永生化细胞系;再用Origene的GFP-tagged FAM83A慢病毒(货号RC208565L4V、RC219879L4V)过表达FAM83A,或用Sigma的shRNA慢病毒(货号TRCN0000168628、TRCN000168368)下调表达。
结果解读:qPCR验证显示,过表达细胞中FAM83A mRNA水平较对照高5–10倍,下调细胞中降低70%以上,模型构建成功。
产品关联:实验所用关键产品:Amsbio的hTERT慢病毒(货号LVP1130)、Origene的GFP-FAM83A慢病毒、Sigma的FAM83A shRNA慢病毒。
3.4 细胞功能实验(活力与增殖)
实验目的:验证FAM83A对乳腺上皮细胞活力与增殖的影响。
方法细节:采用磺酰罗丹明B(SRB)法检测细胞活力(反映代谢状态),溴脱氧尿苷(BrdU)法检测细胞增殖(反映DNA合成);原代与永生化细胞各设3个技术重复,实验重复3次。
结果解读:① 原代细胞:过表达FAM83A后,72小时活力较对照高3.4倍(p>0.05,无统计学意义),但增殖率增加47%(p<0.05);下调FAM83A后,活力降低22%–31%(p=0.003–0.04),但增殖无显著变化。② 永生化细胞:过表达FAM83A显著提升活力(48小时增加90%,72小时增加3.4倍,p<0.05)与增殖(增加40%,p<0.05);下调后两者均显著降低(p<0.05)。
产品关联:实验所用关键试剂:MilliporeSigma的BrdU增殖试剂盒、Thermo Scientific的BCA蛋白定量试剂盒。
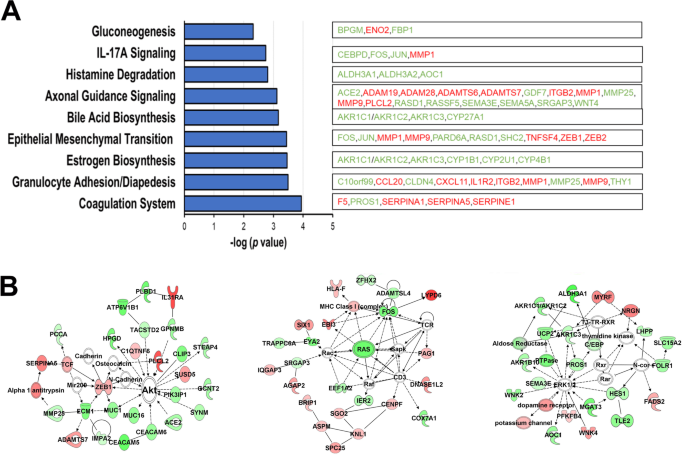
3.5 转录组分析与通路富集
实验目的:解析FAM83A过表达对基因表达的影响。
方法细节:对过表达FAM83A的原代细胞进行RNA-seq(NovaSeq v1.5平台),通过Ingenuity Pathway Analysis(IPA)分析差异表达基因的通路富集。
结果解读:FAM83A过表达显著影响细胞形态、代谢及雌激素生物合成通路:增殖相关基因(如MKI67、PCNA、CCNB1)上调(p<0.05), luminal上皮细胞标记MUC1(参与屏障功能)下调(fold change=0.1,p=0.03),提示细胞向“去分化”方向转变。
3.6 蛋白质相互作用研究
实验目的:鉴定FAM83A的相互作用蛋白,揭示其分子机制。
方法细节:对GFP-tagged FAM83A过表达细胞,用Origene的mGFP抗体磁珠(货号OTI2F6)免疫沉淀,通过Thermo Fisher Eclipse Orbitrap质谱仪分析相互作用蛋白。
结果解读:原代细胞中,FAM83A与DDX3X(转录调控)和LAMB3(细胞粘附)相互作用;永生化细胞中,FAM83A与LIMA1、MYH10、PLEC等细胞骨架重组因子结合。提示FAM83A在不同细胞状态下通过结合不同蛋白发挥功能——原代细胞中参与转录与粘附,永生化细胞中调控细胞骨架重排。
产品关联:实验所用关键产品:Origene的mGFP抗体磁珠(货号OTI2F6)、Thermo Fisher的Eclipse Orbitrap质谱仪。
4. Biomarker研究及发现成果解析
4.1 Biomarker定位与筛选逻辑
FAM83A是乳腺癌起始阶段的潜在蛋白生物标志物,筛选验证逻辑为:
1. 临床组织验证:通过TMA分析正常与肿瘤组织的表达差异;
2. 高风险人群验证:利用公共数据集与独立队列确认易感正常组织中的过表达;
3. 细胞功能验证:在原代与永生化细胞中验证其促增殖作用;
4. 机制解析:通过转录组与蛋白质相互作用研究揭示分子通路。
4.2 研究过程详述
- 临床组织表达:TMA分析显示,肿瘤组织中FAM83A的H-score较正常组织高1.5倍(n=349 vs 411,p<0.0001);且与EGFR表达正相关(正常组织r=0.4,p<0.0001;肿瘤组织r=0.28,p<0.0001)。
- 高风险人群验证:GSE141828数据集显示,易感正常组织(后续确诊乳腺癌)的FAM83A mRNA较健康对照高3.9倍(n=4 vs 8,p=0.02);独立队列中高1.9倍(n=3 vs 8,p=0.03)。
- 细胞功能关联:过表达FAM83A促进原代细胞增殖(增加47%,p<0.05)、永生化细胞活力与增殖(均p<0.05);下调则抑制功能。
- 临床预后价值:肿瘤组织中FAM83A高表达与短生存期相关——阳性率分析显示高表达患者中位生存期缩短(p=0.02),H-score分析结果一致(p=0.04)。
4.3 核心成果提炼
- 早期诊断价值:FAM83A在乳腺癌起始阶段(易感正常组织)已过表达,早于肿瘤形成,是潜在的早期生物标志物;
- 功能意义:促进原代与永生化乳腺上皮细胞增殖,参与代谢激活与细胞骨架重排;
- 机制关联:与EGFR信号通路正相关,可能通过调控EGFR通路启动肿瘤发生;
- 预后价值:肿瘤组织中高表达提示不良预后,可作为预后预测标志物。
综上,本文首次系统证实FAM83A在乳腺癌起始阶段的关键作用,为早期诊断与干预提供了新靶点,也为理解肿瘤“从正常到恶性”的转化机制提供了新视角。