1. 领域背景与文献引入
文献英文标题:AKT phosphorylation as a predictive biomarker for PI3K/mTOR dual inhibition-induced proteolytic cleavage of mTOR companion proteins in small cell lung cancer;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:小细胞肺癌(SCLC)的PI3K/AKT/mTOR通路靶向治疗及生物标志物研究。
小细胞肺癌(SCLC)是肺癌中恶性程度最高的亚型,约占肺癌总数的15%,特点是增殖快、转移早,未经治疗的中位生存期仅2-4个月。自20世纪80年代以来,依托泊苷联合铂类的化疗方案一直是SCLC的标准治疗,但肿瘤易快速复发,广泛期患者5年生存率仅约6%。2019年以来,抗PD-L1抗体(阿替利珠单抗、度伐利尤单抗)联合化疗的方案虽将广泛期患者中位生存期从10.3个月延长至12.3-13.0个月,但获益仍有限,且SCLC目前缺乏明确的靶向治疗策略。
PI3K/AKT/mTOR信号通路是调控细胞生长、存活的关键通路,在SCLC中常因PIK3CA突变、PTEN缺失等发生 constit激活。然而,通路的复杂性(如mTORC1/mTORC2的反馈调节)及肿瘤异质性导致通路抑制剂的治疗响应难以预测——部分携带PIK3CA突变或PTEN缺失的SCLC对PI3K/mTOR抑制剂敏感,但也有肿瘤无响应,临床缺乏有效的预测生物标志物。这一问题严重阻碍了SCLC靶向治疗的精准应用。
针对上述研究空白,本研究聚焦SCLC对PI3K/mTOR双抑制剂的响应机制,探索AKT磷酸化(p-AKT)作为预测生物标志物的价值,并解析双抑制对mTOR伴随蛋白(RICTOR、RPTOR)的水解切割机制,旨在为SCLC的PI3K/mTOR靶向治疗提供伴随诊断依据,解决通路抑制剂响应异质性的临床难题。
2. 文献综述解析
本文综述部分以“PI3K/AKT/mTOR通路在SCLC中的作用→现有通路抑制剂的局限→生物标志物缺乏”为核心逻辑展开评述。现有研究表明,SCLC中PI3K/AKT/mTOR通路激活常见(约30%的肿瘤存在PIK3CA突变或PTEN缺失),但靶向该通路的单药或双抑制剂(如BEZ235、GSK2126458)存在两大局限:一是双抑制剂的严重毒性(如高血糖、腹泻)限制了临床应用;二是肿瘤对抑制剂的响应异质性大,缺乏能预测响应的生物标志物。
现有研究的技术优势在于结合细胞系、动物模型及分子生物学实验(如Western blot、caspase活性检测)解析通路机制,但局限性也较明显:未系统探索SCLC对PI3K/mTOR双抑制剂响应的预测因子,且未明确通路抑制对mTOR伴随蛋白(RICTOR、RPTOR,分别是mTORC2、mTORC1的核心组分)的影响。
本文的创新价值在于:首次明确p-AKT水平与SCLC对PI3K/mTOR双抑制剂的敏感性正相关,且双抑制可通过caspase-6/3介导RICTOR/RPTOR的水解切割;同时提出,p-AKT可作为SCLC患者接受PI3K/mTOR靶向治疗的伴随诊断生物标志物,解决了现有研究中“响应预测困难”的核心问题。
3. 研究思路总结与详细解析
本研究的整体框架为:细胞系筛选→药物敏感性分析→体内验证→机制解析→联合治疗探索,核心目标是明确p-AKT对PI3K/mTOR双抑制剂的预测价值及双抑制诱导mTOR伴随蛋白切割的机制,核心科学问题包括“p-AKT是否可预测SCLC对双抑制剂的响应?”“双抑制如何诱导RICTOR/RPTOR切割?”。
3.1 细胞系PI3K/AKT/mTOR通路激活状态与药物敏感性分析
实验目的:检测13株SCLC细胞系的PI3K/AKT/mTOR通路蛋白表达水平,分析其对不同通路抑制剂的敏感性差异。
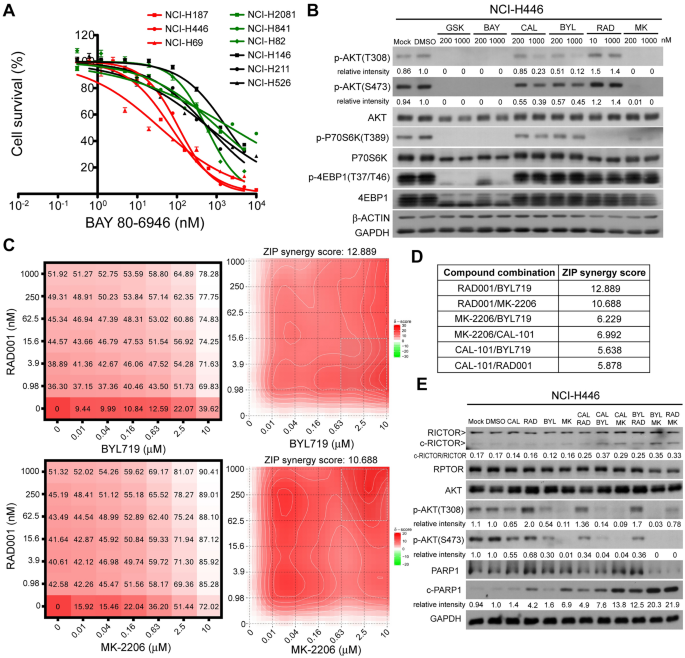
方法细节:选取13株SCLC细胞系(如NCI-H446、NCI-H187、NCI-H211等),通过Western blot检测p-AKT(Thr308/Ser473)、PTEN、PIK3CA、RICTOR、RPTOR等蛋白水平;采用PrestoBlue细胞活力实验,检测细胞对双PI3K/mTOR抑制剂(GSK2126458)、AKT抑制剂(MK-2206)、mTOR抑制剂(RAD001)等的半数抑制浓度(IC50)。
结果解读:p-AKT高表达的细胞系(如NCI-H187、H446、H69,Ser473磷酸化水平>0.7)对GSK2126458高度敏感(IC50<100 nM);而p-AKT低表达的细胞系(如NCI-H211、H524,Ser473磷酸化水平<0.2)对GSK2126458不敏感(IC50>1 μM)。统计分析显示,p-AKT水平与GSK2126458的IC50显著负相关(Thr308:R²=0.581,P=0.0025;Ser473:R²=0.723,P=0.0002),但PTEN水平与敏感性无显著关联。
实验所用关键产品:GSK2126458、MK-2206等试剂购自AdooQ Biosciences、Cayman Chemical;p-AKT、PTEN等抗体购自Cell Signaling Technology;RPTOR、PARP-1抗体购自abcam。
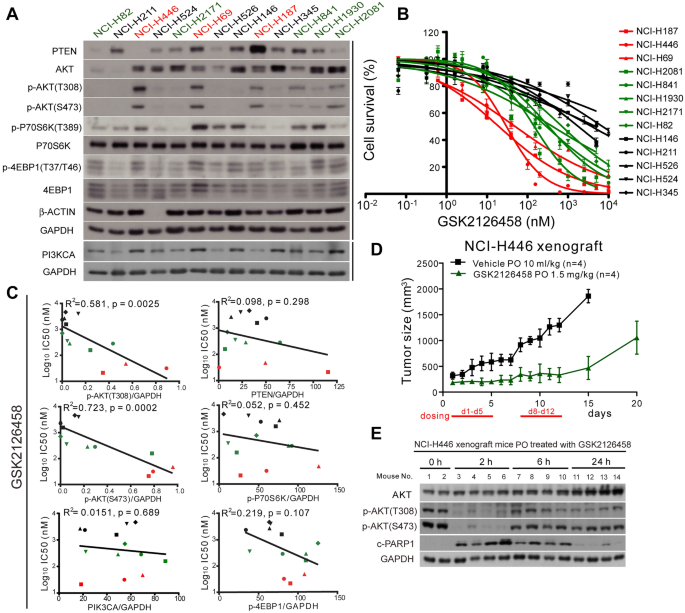
3.2 动物模型验证双抑制剂的体内疗效
实验目的:验证高p-AKT SCLC对PI3K/mTOR双抑制剂的体内响应。
方法细节:将高p-AKT的NCI-H446细胞皮下注射到6-8周雄性裸鼠右侧 flank(1×10⁶细胞/只),待肿瘤体积达150-250 mm³时,随机分为对照组( Vehicle,0.5%甲基纤维素+0.2%吐温-80)和治疗组(GSK2126458,1.5 mg/kg,口服,5天给药2天休息),连续治疗2周。每周测量肿瘤体积(公式:体积=长×宽²/2),治疗结束后取肿瘤组织,通过Western blot检测p-AKT、cleaved PARP-1(凋亡标志物)水平。
结果解读:治疗组肿瘤生长被显著抑制——第12天肿瘤体积较对照组减少>90%(P<0.0001);Western blot显示,给药后2小时肿瘤组织p-AKT(Thr308/Ser473)水平显著降低,cleaved PARP-1水平升高,提示双抑制诱导肿瘤细胞凋亡。
实验所用关键产品:Matrigel基质胶购自Corning;实验动物由BIOLASCO提供。
3.3 PI3K/mTOR双抑制诱导mTOR伴随蛋白切割的机制研究
实验目的:解析PI3K/mTOR双抑制诱导RICTOR、RPTOR水解切割的分子机制。
方法细节:用GSK2126458处理NCI-H446细胞,通过Western blot检测RICTOR、RPTOR的切割产物(c-RICTOR、c-RPTOR);采用siRNA敲低CASP3或CASP6,或用泛caspase抑制剂z-VAD-FMK(10 μM)预处理,观察切割产物的变化;进一步通过重组caspase-3/6实验,验证酶对RICTOR/RPTOR的直接切割作用(细胞裂解液与重组caspase在37℃孵育15-180分钟,Western blot检测切割产物)。
结果解读:GSK2126458处理后,RICTOR被切割为~140 kDa的c-RICTOR(依赖caspase-6),RPTOR被切割为~130 kDa的c-RPTOR(依赖caspase-3);敲低CASP6可显著减少c-RICTOR生成,敲低CASP3可减少c-RPTOR生成,z-VAD-FMK可完全抑制两者切割;重组caspase-6可高效切割RICTOR,caspase-3可高效切割RPTOR,切割产物分子大小与细胞实验一致。
实验所用关键产品:siRNA购自Qiagen(CASP3/6)、ThermoFisher Scientific(PTEN);重组caspase购自abcam;z-VAD-FMK购自AdooQ Biosciences。
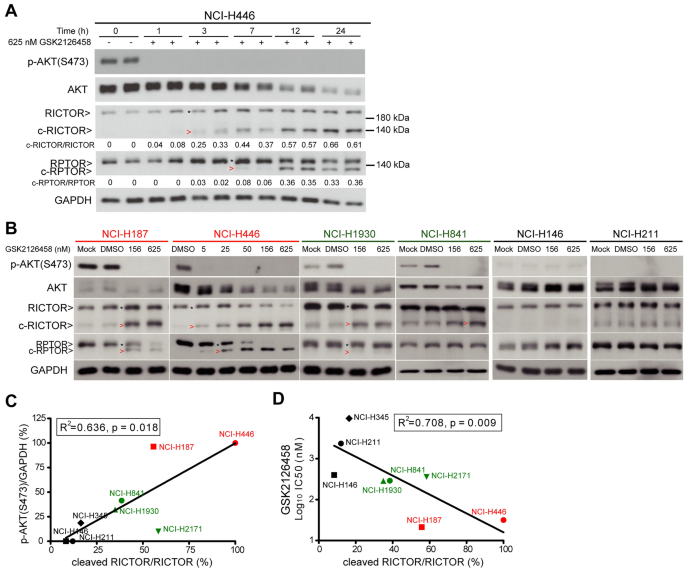
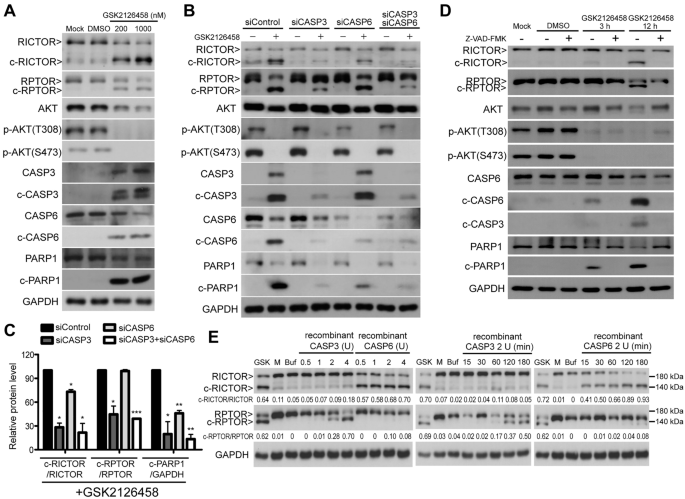
3.4 PTEN缺失对低p-AKT细胞敏感性的影响
实验目的:探索PTEN对低p-AKT SCLC细胞响应双抑制剂的调节作用。
方法细节:选取低p-AKT的NCI-H146、H526细胞,用siRNA敲低PTEN(两种siRNA按1:1混合),48小时后用GSK2126458处理72小时,通过细胞活力实验检测敏感性;Western blot检测p-AKT、c-RICTOR、cleaved PARP-1水平。
结果解读:PTEN敲低后,低p-AKT细胞的p-AKT水平显著升高,对GSK2126458的敏感性增强(IC50降低);同时,c-RICTOR和cleaved PARP-1水平升高,提示PTEN通过调节p-AKT水平影响细胞对双抑制剂的响应。
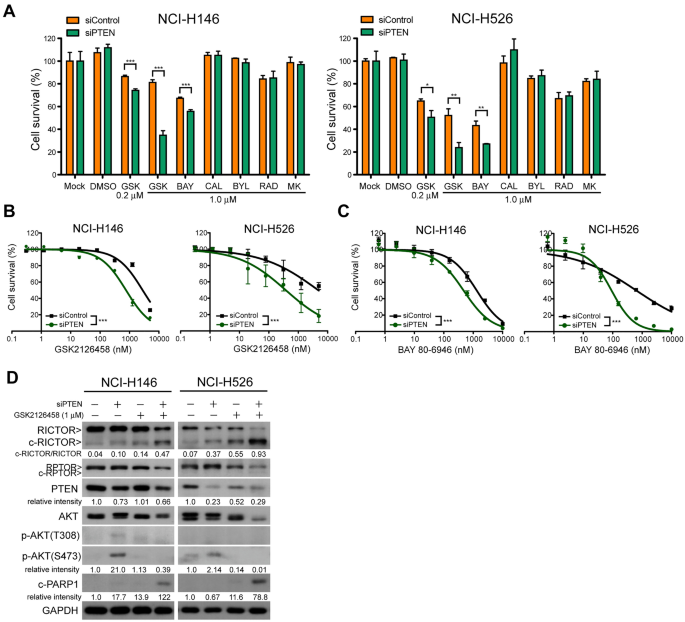
3.5 联合治疗策略探索
实验目的:寻找替代双抑制剂的低毒性联合治疗方案。
方法细节:选取高p-AKT的NCI-H446细胞,采用剂量矩阵法检测PI3Kα选择性抑制剂(BYL719)与mTORC1抑制剂(RAD001)的联合效应;通过SynergyFinder工具(ZIP模型)计算协同分数(>10为协同);Western blot检测联合治疗对p-AKT、c-RICTOR、c-RPTOR、cleaved PARP-1的影响。
结果解读:BYL719与RAD001联合治疗的协同分数为12.889(显著协同),可显著降低p-AKT水平,诱导c-RICTOR/c-RPTOR生成及cleaved PARP-1升高,细胞凋亡率高于单药治疗。
实验所用关键产品:BYL719购自Cayman Chemical;RAD001购自AdooQ Biosciences。
4. Biomarker研究及发现成果解析
Biomarker定位与筛选逻辑
本研究的核心生物标志物是AKT磷酸化水平(p-AKT(Thr308/Ser473)),其筛选与验证遵循“细胞系关联分析→体内模型验证→功能调控实验”的完整逻辑:首先通过13株SCLC细胞系的药物敏感性分析,发现p-AKT与双抑制剂IC50显著负相关;随后通过动物模型验证高p-AKT肿瘤对双抑制剂敏感;最后通过PTEN敲低实验,确认p-AKT是调节响应的关键因子。
研究过程详述
Biomarker来源:SCLC细胞系及异种移植瘤组织的蛋白表达(通过Western blot检测)。
验证方法:① 细胞系层面:13株细胞系的p-AKT水平与GSK2126458的IC50进行Pearson相关性分析,结果显示Ser473位点磷酸化的相关性最强(R²=0.723,P=0.0002);② 体内层面:高p-AKT的NCI-H446异种移植瘤对GSK2126458敏感(肿瘤抑制率>90%);③ 功能层面:PTEN敲低(升高p-AKT)可增强低p-AKT细胞对双抑制剂的敏感性。
特异性与敏感性:p-AKT(Ser473)>0.7的细胞系对GSK2126458的敏感性(IC50<100 nM)显著高于p-AKT(Ser473)<0.2的细胞系(IC50>1 μM),提示p-AKT具有良好的预测特异性与敏感性(文献未提供ROC曲线数据,但相关性分析具有统计学意义)。
核心成果提炼
- 预测价值:p-AKT水平可作为SCLC对PI3K/mTOR双抑制剂响应的预测生物标志物——高p-AKT肿瘤更敏感,低p-AKT肿瘤不敏感(Ser473位点相关性:R²=0.723,P=0.0002)。
- 机制创新:首次发现PI3K/mTOR双抑制可诱导mTOR伴随蛋白的水解切割——RICTOR依赖caspase-6切割,RPTOR依赖caspase-3切割,且切割程度与p-AKT水平正相关(R²=0.636,P=0.018)。
- 临床意义:提出高p-AKT的SCLC患者可从“PI3Kα抑制剂+ mTORC1抑制剂”联合治疗中获益,为临床规避双抑制剂的毒性提供了替代方案;同时,p-AKT可作为伴随诊断标志物,筛选适合PI3K/mTOR靶向治疗的患者。
本研究通过系统的细胞、动物及分子实验,明确了p-AKT作为SCLC PI3K/mTOR靶向治疗预测生物标志物的价值,解析了双抑制诱导mTOR伴随蛋白切割的机制,为SCLC的精准靶向治疗提供了重要的理论与实验依据。