1. 领域背景与文献引入
文献英文标题:Draft genome sequence of the Daphnia pathogen Octosporea bayeri: insights into the gene content of a large microsporidian genome and a model for host-parasite interactions;发表期刊:Genome Biology;影响因子:未公开(2009年Genome Biology影响因子约10.0);研究领域:微孢子虫基因组学、宿主-寄生虫相互作用。
微孢子虫是一类高度适应专性细胞内寄生的真核生物,可感染节肢动物、鱼类、哺乳动物(包括人类),其核心特征是具有用于宿主细胞入侵的“极丝”结构。早期研究认为微孢子虫是“原始真核生物”(无线粒体、高尔基体,含70S核糖体),但后续基因组与细胞生物学研究证实:这些特征是寄生生活导致的极端简化结果——微孢子虫实际与真菌亲缘关系密切,具有退化的线粒体(称为“mitosomes”)。
已测序的微孢子虫基因组多为小基因组类群(如Encephalitozoon cuniculi,仅2.9Mb,约2000个基因),其特点是基因紧凑(基因间区短)、代谢途径严重简化(缺失嘌呤/嘧啶从头合成、三羧酸循环等关键途径),高度依赖宿主提供能量与代谢物。然而,微孢子虫基因组大小差异可达6倍(2.9-24Mb),大基因组类群的研究仅局限于小规模序列调查(如Spraguea lophii的EST分析),无法回答“基因组大小差异源于基因数量增加还是非编码序列扩张”这一核心问题。
本研究针对“小基因组无法代表所有微孢子虫、大基因组研究不深入”的空白,首次对大基因组微孢子虫Octosporea bayeri(感染水蚤Daphnia magna,基因组约24Mb)进行深度测序与分析,旨在揭示大基因组的结构特征、基因内容及进化意义,为微孢子虫的基因组演化研究提供新模型。
2. 文献综述解析
作者对微孢子虫现有研究的分类维度为“基因组大小”,将研究分为两类:
1. 小基因组微孢子虫(2.9-6Mb):以Enc. cuniculi、Enterocytozoon bieneusi为代表,研究证实其基因组紧凑、基因数量少(约2000个)、代谢途径简化,高度依赖宿主;
2. 大基因组微孢子虫(>10Mb):仅小规模序列调查(如Vittaforma corneae的部分基因组),显示非编码DNA比例高,但无法了解整体结构与基因内容。
现有研究的局限性:
- 小基因组类群的研究无法代表所有微孢子虫;
- 大基因组类群的研究样本量小、覆盖度低,无法回答“基因组大小差异的机制”。
本研究的创新价值:
首次通过深度Illumina测序(覆盖度34.2-37.2×)解析大基因组微孢子虫的基因组,揭示其基因密度高度不均(存在基因密集区与长非编码区)、基因内容更复杂(比小基因组多80个功能基因)、代谢途径更完整(减少对宿主的依赖),修正了“微孢子虫基因组均为小而紧凑”的传统认知。
3. 研究思路总结与详细解析
本研究以“揭示大基因组微孢子虫的结构与功能”为目标,采用“基因组测序→基因注释→比较分析→结构解析”的闭环路线,从序列、基因、功能、进化多层面展开研究。
3.1 基因组DNA提取与测序
实验目的:获取O. bayeri的高质量基因组DNA并完成测序,为后续组装提供基础。
方法细节:从实验室培养的感染D. magna中纯化O. bayeri孢子,提取基因组DNA(100ng);使用Illumina Genome Analyzer进行35bp单端/配对端测序(由FASTERIS SA完成);通过EDENA、Velvet、ELAND软件组装序列。
结果解读:生成898Mb原始序列,组装为41804个重叠群(contig),总长度13.3Mb(GC含量26%);通过覆盖度估算基因组大小约24Mb(已知最大的微孢子虫基因组);仅20个contig存在污染,其余为O. bayeri序列。
产品关联:实验设备为Illumina Genome Analyzer(FASTERIS SA提供测序服务);未提及具体DNA提取试剂盒,领域常规使用Qiagen DNeasy等商业化试剂盒。
3.2 基因注释与同源性分析
实验目的:鉴定O. bayeri的基因(rRNA、tRNA、蛋白编码基因),并对比已知微孢子虫的基因内容。
方法细节:
- 用TBLASTX对比Enc. cuniculi基因组(E≤1e-10),鉴定同源基因;
- 用EMBOSS GETORF预测开放阅读框(ORF),再用BLASTP对比NCBI非冗余数据库(E≤1e-10),鉴定真核同源基因;
- 用tRNAscan-SE鉴定tRNA基因。
结果解读:
- 注释得到4个rRNA、37个tRNA、2174个蛋白编码ORF;
- 1405个ORF与Enc. cuniculi同源(占Enc. cuniculi基因的70%),其中93%的功能基因与53%的假设基因在O. bayeri中存在;
- 80个ORF与真核生物(主要是真菌)同源但不在Enc. cuniculi中,涉及转录(如RNA聚合酶)、代谢(如脂肪酸酶)等功能;
- 689个ORF无同源性(25个与Antonospora locustae的假设蛋白同源)。
产品关联:使用NCBI BLASTALL、EMBOSS GETORF等工具,未提及具体品牌,领域常规使用这些工具。
3.3 系统发育与功能分类分析
实验目的:确定O. bayeri的进化位置,对比小基因组微孢子虫的功能差异。
方法细节:
- 系统发育:收集13种微孢子虫的α/β-tubulin序列,用Muscle对齐、Gblocks筛选保守区,MrBayes构建进化树(GTR模型);
- 功能分类:将O. bayeri的ORF按Enc. cuniculi的11个功能类别(代谢、能量产生等)分类,对比Enc. cuniculi、Ent. bieneusi的差异。
结果解读:
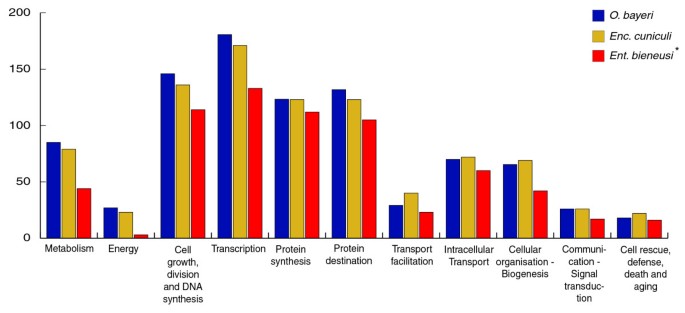
- 进化树显示O. bayeri位于微孢子虫基部,与大基因组类群(如Brachiola algerae)亲缘近,小基因组类群(Enc. cuniculi、Nosema ceranae)形成独立分支;
- 功能分类显示O. bayeri的代谢、能量产生基因数量多于小基因组类群(如脂质代谢基因37个vs Enc. cuniculi的29个),而转运蛋白数量更少(如ATP转运蛋白1个vs Enc. cuniculi的4个)。
《Draft genome sequence of the Daphnia pathogen Octosporea bayeri: insights into the gene content of a large microsporidian genome and a model for host-parasite interactions》-文献解析
1. 领域背景与文献引入
文献英文标题:Draft genome sequence of the Daphnia pathogen Octosporea bayeri: insights into the gene content of a large microsporidian genome and a model for host-parasite interactions;发表期刊:Genome Biology;影响因子:未公开(2009年Genome Biology影响因子约10.0);研究领域:微孢子虫基因组学、宿主-寄生虫相互作用。
微孢子虫是一类高度适应专性细胞内寄生的真核生物,可感染节肢动物、鱼类、哺乳动物(包括人类),其核心特征是具有用于宿主细胞入侵的“极丝”结构。早期研究认为微孢子虫是“原始真核生物”(无线粒体、高尔基体,含70S核糖体),但后续基因组与细胞生物学研究证实:这些特征是寄生生活导致的极端简化结果——微孢子虫实际与真菌亲缘关系密切,具有退化的线粒体(称为“mitosomes”)。
已测序的微孢子虫基因组多为小基因组类群(如Encephalitozoon cuniculi,仅2.9Mb,约2000个基因),其特点是基因紧凑(基因间区短)、代谢途径严重简化(缺失嘌呤/嘧啶从头合成、三羧酸循环等关键途径),高度依赖宿主提供能量与代谢物。然而,微孢子虫基因组大小差异可达6倍(2.9-24Mb),大基因组类群的研究仅局限于小规模序列调查(如Spraguea lophii的EST分析),无法回答“基因组大小差异源于基因数量增加还是非编码序列扩张”这一核心问题。
本研究针对“小基因组无法代表所有微孢子虫、大基因组研究不深入”的空白,首次对大基因组微孢子虫Octosporea bayeri(感染水蚤Daphnia magna,基因组约24Mb)进行深度测序与分析,旨在揭示大基因组的结构特征、基因内容及进化意义,为微孢子虫的基因组演化研究提供新模型。
2. 文献综述解析
作者对微孢子虫现有研究的分类维度为“基因组大小”,将研究分为两类:
1. 小基因组微孢子虫(2.9-6Mb):以Enc. cuniculi、Enterocytozoon bieneusi为代表,研究证实其基因组紧凑、基因数量少(约2000个)、代谢途径简化,高度依赖宿主;
2. 大基因组微孢子虫(>10Mb):仅小规模序列调查(如Vittaforma corneae的部分基因组),显示非编码DNA比例高,但无法了解整体结构与基因内容。
现有研究的局限性:
- 小基因组类群的研究无法代表所有微孢子虫;
- 大基因组类群的研究样本量小、覆盖度低,无法回答“基因组大小差异的机制”。
本研究的创新价值:
首次通过深度Illumina测序(覆盖度34.2-37.2×)解析大基因组微孢子虫的基因组,揭示其基因密度高度不均(存在基因密集区与长非编码区)、基因内容更复杂(比小基因组多80个功能基因)、代谢途径更完整(减少对宿主的依赖),修正了“微孢子虫基因组均为小而紧凑”的传统认知。
3. 研究思路总结与详细解析
本研究以“揭示大基因组微孢子虫的结构与功能”为目标,采用“基因组测序→基因注释→比较分析→结构解析”的闭环路线,从序列、基因、功能、进化多层面展开研究。
3.1 基因组DNA提取与测序
实验目的:获取O. bayeri的高质量基因组DNA并完成测序,为后续组装提供基础。
方法细节:从实验室培养的感染D. magna中纯化O. bayeri孢子,提取基因组DNA(100ng);使用Illumina Genome Analyzer进行35bp单端/配对端测序(由FASTERIS SA完成);通过EDENA、Velvet、ELAND软件组装序列。
结果解读:生成898Mb原始序列,组装为41804个重叠群(contig),总长度13.3Mb(GC含量26%);通过覆盖度估算基因组大小约24Mb(已知最大的微孢子虫基因组);仅20个contig存在污染,其余为O. bayeri序列。
产品关联:实验设备为Illumina Genome Analyzer(FASTERIS SA提供测序服务);未提及具体DNA提取试剂盒,领域常规使用Qiagen DNeasy等商业化试剂盒。
3.2 基因注释与同源性分析
实验目的:鉴定O. bayeri的基因(rRNA、tRNA、蛋白编码基因),并对比已知微孢子虫的基因内容。
方法细节:
- 用TBLASTX对比Enc. cuniculi基因组(E≤1e-10),鉴定同源基因;
- 用EMBOSS GETORF预测开放阅读框(ORF),再用BLASTP对比NCBI非冗余数据库(E≤1e-10),鉴定真核同源基因;
- 用tRNAscan-SE鉴定tRNA基因。
结果解读:
- 注释得到4个rRNA、37个tRNA、2174个蛋白编码ORF;
- 1405个ORF与Enc. cuniculi同源(占Enc. cuniculi基因的70%),其中93%的功能基因与53%的假设基因在O. bayeri中存在;
- 80个ORF与真核生物(主要是真菌)同源但不在Enc. cuniculi中,涉及转录(如RNA聚合酶)、代谢(如脂肪酸酶)等功能;
- 689个ORF无同源性(25个与Antonospora locustae的假设蛋白同源)。
产品关联:使用NCBI BLASTALL、EMBOSS GETORF等工具,未提及具体品牌,领域常规使用这些工具。
3.3 系统发育与功能分类分析
实验目的:确定O. bayeri的进化位置,对比小基因组微孢子虫的功能差异。
方法细节:
- 系统发育:收集13种微孢子虫的α/β-tubulin序列,用Muscle对齐、Gblocks筛选保守区,MrBayes构建进化树(GTR模型);
- 功能分类:将O. bayeri的ORF按Enc. cuniculi的11个功能类别(代谢、能量产生等)分类,对比Enc. cuniculi、Ent. bieneusi的差异。
结果解读:
- 进化树显示O. bayeri位于微孢子虫基部,与大基因组类群(如Brachiola algerae)亲缘近,小基因组类群(Enc. cuniculi、Nosema ceranae)形成独立分支;
- 功能分类显示O. bayeri的代谢、能量产生基因数量多于小基因组类群(如脂质代谢基因37个vs Enc. cuniculi的29个),而转运蛋白数量更少(如ATP转运蛋白1个vs Enc. cuniculi的4个)。
3.4 基因密度与非编码序列分析
实验目的:解析O. bayeri基因组的基因密度分布,探究大基因组的结构特征。
方法细节:选取200个最长的contig(平均长度2795bp),注释长度≥100aa的ORF,计算基因密度(基因间区长度);通过覆盖度分析重复序列(高覆盖度contig为重复区)。
结果解读:
- 基因密度高度不均:50%的contig无ORF(长非编码区),部分contig基因间区仅429bp(比Enc. cuniculi更紧凑);整体基因密度为1基因/4593bp(是小基因组的1/5);
- 重复序列分析:高覆盖度contig(>200×)均为短序列(<300bp),提示存在大量重复元件(如Mariner、Gypsy转座子);1345个contig含串联重复序列(多为短串联重复)。
产品关联:使用Tandem Repeat Finder分析串联重复,领域常规工具。
3.5 内含子与蛋白长度分析
实验目的:对比小基因组微孢子虫,分析O. bayeri的内含子特征与蛋白长度差异。
方法细节:手动筛查ORF中的移码突变(潜在内含子),对比Enc. cuniculi的内含子位置;选取完整ORF,对比O. bayeri与Enc. cuniculi、真菌(如S. cerevisiae)的蛋白长度。
结果解读:
- 内含子:仅鉴定6个内含子,均位于核糖体蛋白基因(如L19、L27a)的起始密码子附近,与Enc. cuniculi的内含子位置一致;
- 蛋白长度:O. bayeri的蛋白比Enc. cuniculi长3%(69%的蛋白更长),但比真菌短14%(75%的蛋白更短),提示微孢子虫蛋白整体简化,但大基因组蛋白保留更多结构域。
3.6 ATP转运蛋白的进化分析
实验目的:探究O. bayeri的能量获取方式,对比小基因组的宿主依赖差异。
方法细节:鉴定O. bayeri的ATP转运蛋白基因,用MrBayes构建进化树(对比其他微孢子虫的ATP转运蛋白)。
结果解读:
- O. bayeri仅1个ATP转运蛋白,进化树显示其为微孢子虫ATP转运蛋白的基部类群;
- 结合功能分类结果(O. bayeri的转运蛋白数量少、代谢基因多),推测O. bayeri对宿主的能量依赖弱于小基因组类群。
4. Biomarker研究及发现成果解析
本研究未涉及Biomarker筛选或验证,主要聚焦于基因组结构与进化分析。但从研究结果推测,O. bayeri的代谢基因(如脂质代谢酶)或ATP转运蛋白可能作为大基因组微孢子虫的“特征基因”,为后续微孢子虫的分类与诊断提供潜在靶点(推测:这些基因可作为大基因组微孢子虫的分子标记,需临床样本验证)。
此外,O. bayeri与宿主D. magna的基因组互补(D. magna基因组已测序),为宿主-寄生虫相互作用研究提供了理想模型——后续可通过转录组分析,鉴定O. bayeri感染后D. magna的差异表达基因,筛选调控寄生的关键因子(潜在Biomarker)。