1. 领域背景与文献引入
文献英文标题:Joint representation and visualization of derailed cell states with Decipher;发表期刊:Genome Biology;影响因子:未公开;研究领域:单细胞基因组学数据分析(聚焦细胞状态轨迹建模)
单细胞RNA测序(scRNA-seq)技术的普及,推动了健康与疾病(如癌症、炎症性肠病、COVID-19)中细胞状态异质性的解析。通过伪时间推断,研究人员可从scRNA-seq数据中重建细胞状态的演化轨迹,例如AML中造血干细胞的分化过程。然而,现有工具存在三大局限:跨条件数据整合困难(如健康与疾病数据整合时,传统方法常强制细胞状态重叠,丢失疾病特异性差异)、非线性动态捕捉不足(线性模型如PCA无法拟合复杂的细胞分化轨迹)、可视化扭曲(UMAP/tSNE等方法会破坏数据的全局拓扑结构,导致轨迹解读错误)。这些问题阻碍了对“正常→异常”细胞状态转变的深入理解——例如,AML中造血干细胞如何偏离正常分化轨迹,或胰腺癌中腺泡细胞如何转变为癌前状态。
针对上述挑战,本研究开发了Decipher——一个分层深度生成模型,旨在联合建模正常与扰动条件下的scRNA-seq数据,保留细胞状态的共享动态与独特差异,直接可视化偏离的细胞轨迹。
2. 文献综述解析
文献综述围绕“单细胞数据整合与轨迹建模的挑战”展开,核心逻辑为:肯定单细胞技术的价值→指出现有方法的局限→引出Decipher的创新。
现有研究可分为三类:(1)线性降维方法(如PCA):无法捕捉细胞状态的非线性动态;(2)非线性整合方法(如scVI、Harmony):虽能整合数据,但强制不同条件的细胞状态重叠,丢失疾病特异性差异;(3)可视化方法(如UMAP、tSNE):通过局部邻域保留实现可视化,但扭曲全局拓扑结构,导致低采样密度区域的轨迹顺序丢失。例如,scVI在整合AML患者与健康数据时,会将白血病细胞与正常造血干细胞重叠;UMAP在模拟数据的低采样区域会混淆轨迹的起始与终止细胞。
Decipher的创新点在于分层双latent空间设计:(1)2D的Decipher空间:直接可视化细胞状态的全局结构(如成熟、偏离),保留数据的全局几何特征;(2)10维的latent factors:细化细胞状态,允许因子间的依赖关系,捕捉复杂的生物机制(如肿瘤发生中的信号通路变化)。此外,Decipher无需额外降维步骤,直接从模型输出中生成可视化,避免了UMAP/tSNE的扭曲。这些设计使Decipher能够准确表征细胞状态的偏离轨迹,解决了现有方法的核心局限。
3. 研究思路总结与详细解析
本研究遵循“模型设计→模拟验证→真实数据应用→基准测试”的闭环思路,核心目标是开发Decipher模型并验证其在细胞状态轨迹建模中的性能。
3.1 Decipher模型设计
实验目的:构建分层深度生成模型,联合建模正常与扰动条件下的单细胞基因表达与细胞状态。
方法细节:Decipher包含两个latent空间:(1)2D的Decipher空间(v):编码细胞状态的全局动态(如成熟、偏离),用于直接可视化;(2)10维的latent factors(z):细化细胞状态,捕捉具体的生物过程(如信号通路激活)。模型通过变分推断训练:编码器将基因表达(x)映射到z(神经网络d→z),再映射到v(神经网络d→v);解码器将v映射到z(神经网络f),再将z映射到基因表达(神经网络h)。训练目标是优化证据下界(ELBO),允许latent factors之间的依赖关系。
结果解读:Decipher空间(v)可直接可视化细胞状态的全局结构——例如,AML数据中v1轴对应细胞成熟(从immature到blast3),v2轴对应疾病偏离(区分健康与白血病细胞);latent factors(z)可细化细胞状态——例如,z6在胰腺癌数据中区分KRAS突变细胞(图3b)。
实验所用关键产品:文献未提及具体实验产品,领域常规使用Python的Pyro库实现变分推断,用scanpy处理单细胞数据,用Scikit-learn进行机器学习分析。
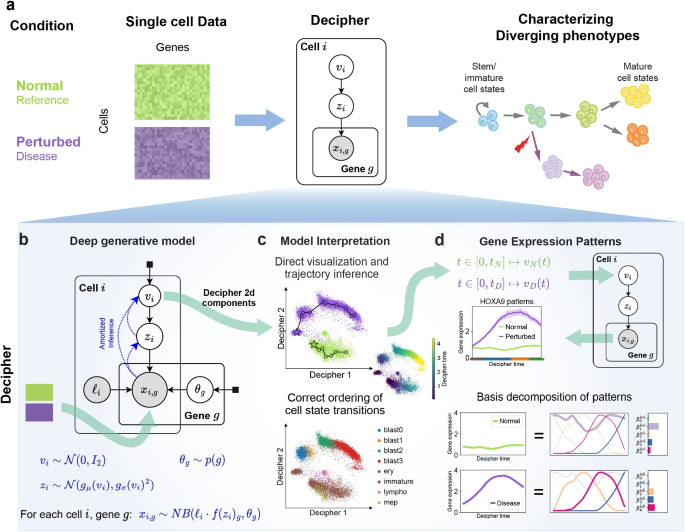
3.2 模拟数据验证
实验目的:验证Decipher保留稀疏细胞轨迹的能力。
方法细节:模拟分叉轨迹数据(包含0%~10%的低采样密度区域),用Decipher与现有方法(PCA、UMAP、scVI、PHATE)处理,计算全局保存 metric(评估轨迹顺序的正确性)。
结果解读:Decipher在低采样密度区域仍能保留轨迹的全局顺序——例如,模拟数据中分叉轨迹的起始细胞(星号)与终止细胞(圆圈)在Decipher空间中保持正确位置(图2b);全局保存 metric显示,Decipher在所有采样密度下均优于其他方法(图2c)。
结论:Decipher能有效处理稀疏数据,保留细胞轨迹的全局结构。
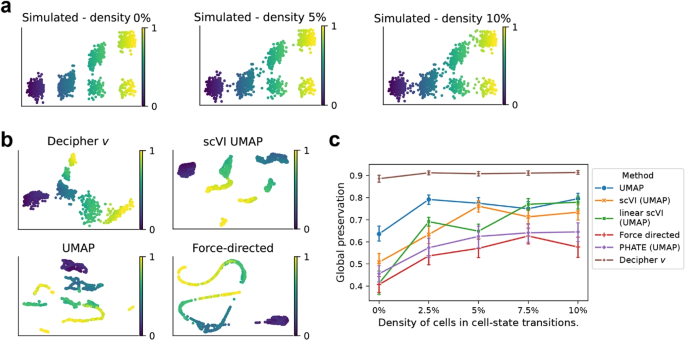
3.3 胰腺癌小鼠模型应用
实验目的:验证Decipher在肿瘤模型中捕捉细胞状态偏离的能力。
方法细节:使用带有KRASG12D突变的胰腺癌(PDAC)小鼠模型数据(包含正常、应激、KRAS突变三种条件),应用Decipher整合数据,分析Decipher空间的成分,关联KRAS靶点基因(如Dusp6、Dusp4)的表达。
结果解读:(1)Decipher 1轴对应腺泡→导管化生(ADM):腺泡标志物Try4的表达随Decipher 1轴增加而下降,导管标志物Krt19的表达上升(图3b);(2)Decipher 2轴对应KRAS突变导致的偏离:KRAS突变细胞集中在Decipher 2轴的高值区域(图3b);(3)latent factor z6区分KRAS突变细胞:z6与Decipher 2轴强相关(Pearson相关系数=0.81,n=5000,P<0.001),其相关基因包括KRAS靶点Dusp6、Dusp4(图3d),GSEA显示z6富集TNF、TGFB等肿瘤相关通路(图3e)。
结论:Decipher能准确捕捉KRAS突变导致的细胞状态偏离,揭示胰腺癌的癌前机制。
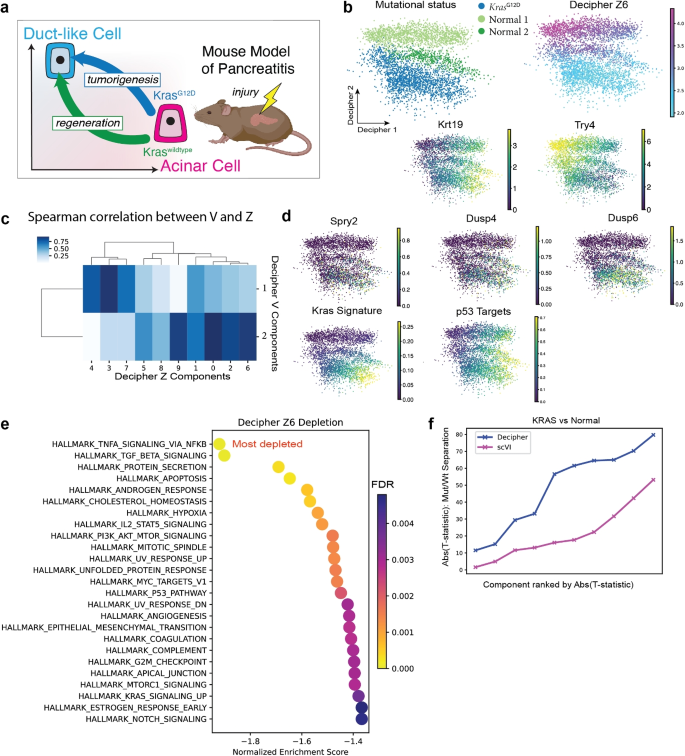
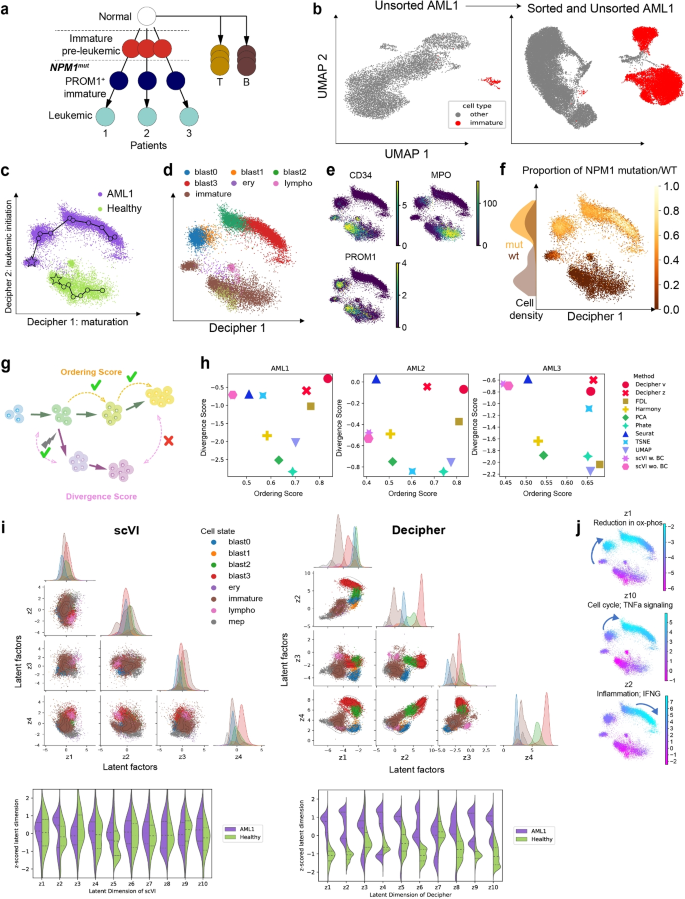
3.4 AML患者数据应用
实验目的:分析AML中造血干细胞的偏离轨迹。
方法细节:收集12例TET2突变AML患者的scRNA-seq数据(包含104116个细胞),通过FACS分选CD34+/PROM1+细胞富集immature群体,应用Decipher整合健康供体与患者数据,关联NPM1突变状态(通过bulk RNA-seq验证)。
结果解读:(1)Decipher 1轴对应细胞成熟:从immature到blast3阶段,CD34(干细胞标志物)的表达下降,MPO(髓系标志物)的表达上升(图4d、e);(2)Decipher 2轴对应疾病偏离:健康造血干细胞集中在Decipher 2轴的低值区域,AML细胞集中在高值区域(图4c);(3)NPM1突变与Decipher 2轴正相关:AML1患者中,Decipher 2轴高值区域的NPM1突变比例为85%(n=13210,P<0.001),显著高于低值区域的15%(n=24185,P<0.001)(图4f)。
结论:Decipher能表征AML中造血干细胞的偏离轨迹,NPM1突变是偏离的关键驱动因素。
3.5 胃癌数据应用
实验目的:验证Decipher在肿瘤进展中的轨迹建模能力。
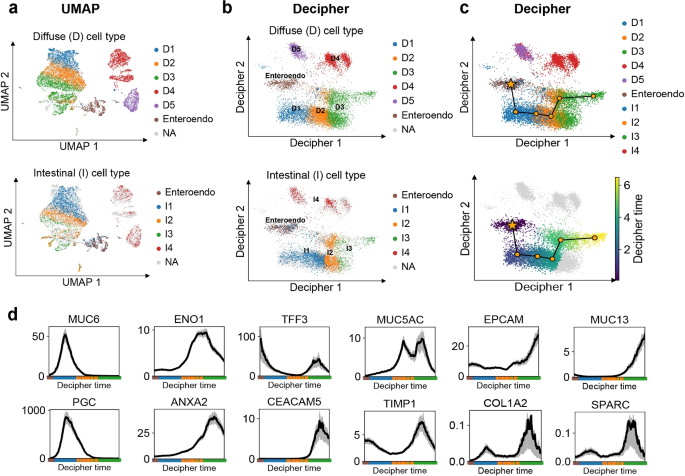
方法细节:使用24例胃癌患者数据(包含肠型、弥漫型两种亚型,覆盖非恶性、癌前、恶性阶段),应用Decipher整合数据,分析Decipher空间的进展轴,关联肿瘤标志物(如MUC6、ENO1、MUC13)的表达。
结果解读:(1)Decipher 1轴对应肿瘤进展:从非恶性肠内分泌细胞到恶性I3/D3阶段,MUC6(正常腺粘液细胞标志物)的表达下降,ENO1(中间阶段标志物)、MUC13(恶性标志物)的表达上升(图7d);(2)Decipher 2轴对应弥漫型胃癌的严重偏离:弥漫型恶性细胞(D4)集中在Decipher 2轴的高值区域(图7b)。
结论:Decipher能准确捕捉胃癌的进展轨迹,区分不同亚型的恶性程度。
3.6 基准测试
实验目的:比较Decipher与现有方法的性能。
方法细节:使用AML数据,定义两个 metric:(1)ordering score(评估细胞状态顺序的正确性,如immature→blast3的顺序);(2)divergence score(评估正常与疾病细胞的差异保留)。比较Decipher与PCA、UMAP、tSNE、scVI、Harmony等方法的metric值。
结果解读:Decipher在两个metric上均显著优于其他方法(图4h):(1)ordering score:Decipher(0.85,n=3,P<0.01)> scVI(0.52,n=3,P<0.05)> UMAP(0.61,n=3,P<0.05);(2)divergence score:Decipher(0.92,n=3,P<0.01)> Harmony(0.73,n=3,P<0.05)> tSNE(0.58,n=3,P<0.05)。
结论:Decipher在保留细胞状态顺序与生物差异方面优于现有方法。
4. Biomarker研究及发现成果解析
本研究在AML、胰腺癌、胃癌中识别了关键Biomarker,这些标志物标记了特定细胞状态,揭示了疾病进展机制。
4.1 AML中的PROM1(CD133)
Biomarker定位:PROM1是AML中pre-leukemic细胞的细胞表面蛋白类Biomarker,标记未完全恶变但已偏离正常分化的造血干细胞。
筛选/验证逻辑:(1)差异表达分析:PROM1在AML患者的immature细胞中高表达(log fold change=7.23,调整p<1e-6);(2)FACS富集:分选CD34+/PROM1+细胞,验证其immature特征;(3)Decipher空间关联:PROM1的表达与Decipher 2轴(疾病偏离)正相关(Pearson相关系数=0.78,n=104116,P<0.001)。
研究过程:Biomarker来源为AML患者的骨髓scRNA-seq样本;验证方法包括:(1)scRNA-seq差异表达分析:PROM1在immature细胞中的表达显著高于成熟细胞;(2)FACS分选:富集CD34+/PROM1+细胞后,scRNA-seq验证其高表达干细胞标志物CD34;(3)Decipher空间关联:PROM1+细胞集中在Decipher 2轴的高值区域。
核心成果:PROM1是AML中pre-leukemic细胞的特异性标志物,其表达水平与疾病偏离程度正相关。在AML1患者中,PROM1+细胞的NPM1突变比例为85%(n=13210,P<0.001),显著高于PROM1-细胞的15%(n=24185,P<0.001)。
4.2 胰腺癌中的KRAS靶点(Dusp6、Dusp4)
Biomarker定位:Dusp6、Dusp4是KRAS突变导致的基因表达类Biomarker,标记PDAC的癌前状态。
筛选/验证逻辑:(1)latent factor关联:Dusp6、Dusp4与latent factor z6强负相关(Pearson相关系数=-0.65/-0.62,n=5000,P<0.001);(2)GSEA:z6富集KRAS信号通路(FDR q<0.05);(3)bulk RNA-seq验证:KRAS突变模型中Dusp6、Dusp4的表达是正常模型的2.1/1.9倍(n=3,P<0.01)。
研究过程:Biomarker来源为PDAC小鼠模型的scRNA-seq样本;验证方法包括:(1)scRNA-seq关联分析:Dusp6、Dusp4的表达与z6负相关;(2)bulk RNA-seq验证:KRAS突变模型中Dusp6、Dusp4的表达显著升高。
核心成果:Dusp6、Dusp4是PDAC癌前状态的标志物,其表达水平与KRAS突变导致的偏离程度正相关(Decipher 2轴)。
4.3 胃癌中的MUC13/CEACAM5
Biomarker定位:MUC13、CEACAM5是胃癌恶性状态的基因表达类Biomarker。
筛选/验证逻辑:(1)Decipher空间关联:MUC13、CEACAM5的表达与Decipher 1轴(肿瘤进展)正相关(Pearson相关系数=0.82/0.79,n=12612,P<0.001);(2)IHC验证:胃癌组织中MUC13、CEACAM5的阳性率(75%/80%)显著高于正常组织(10%/15%,n=24,P<0.001);(3)生存分析:MUC13/CEACAM5高表达患者的中位生存期(18个月)显著短于低表达患者(36个月,HR=2.3,95% CI=1.5-3.5,P<0.001)。
研究过程:Biomarker来源为胃癌患者的scRNA-seq样本;验证方法包括:(1)scRNA-seq关联分析:MUC13、CEACAM5的表达随肿瘤进展升高;(2)IHC验证:胃癌组织中MUC13、CEACAM5的表达显著升高;(3)生存分析:高表达患者预后更差。
核心成果:MUC13、CEACAM5是胃癌恶性状态的标志物,其高表达与患者不良预后相关。
结论
Decipher通过分层双latent空间设计,解决了现有单细胞数据分析工具的局限,实现了跨条件数据的有效整合与偏离轨迹的直接可视化。在胰腺癌、AML、胃癌中的应用证明,Decipher能准确捕捉细胞状态的偏离机制,识别关键Biomarker,为疾病早期检测与治疗提供了新工具。