1. 领域背景与文献引入
文献英文标题:Regulatory analysis of single cell multiome gene expression and chromatin accessibility data with scREG;发表期刊:Genome Biology;影响因子:未公开;研究领域:单细胞多组学整合分析(基因表达与染色质可及性联合调控研究)
领域共识:单细胞测序技术的发展推动了生命科学研究的精细化,单细胞RNA测序可解析细胞转录组异质性,单细胞转座酶可及性测序可揭示染色质开放区域的调控特征。传统研究中,两种组学数据通常来自异质群体的不同细胞亚样本,现有整合方法依赖基因组距离或外部数据建立顺式调控元件与靶基因的关联,无法直接利用同一细胞内两种组学的关联信息,导致顺式调控推断能力受限。近年来,同一细胞内联合检测基因表达与染色质可及性的多组学技术(如10X Genomics Chromium平台)逐渐普及,为直接解析细胞内调控关联提供了数据基础,但缺乏能充分利用此类数据的整合分析方法。本文针对这一研究空白,开发了基于顺式调控潜能的scREG方法,实现单细胞多组学数据的联合降维、细胞分群与亚群特异性调控网络构建,为疾病关联位点的解析提供新工具。
2. 文献综述解析
作者对领域内现有单细胞多组学整合方法的分类维度为顺式调控元件与靶基因的链接函数类型,主要分为三类:基于基因组距离的方法、基于外部数据的方法、未充分利用同一细胞关联信息的联合分析方法。
现有研究中,基于基因组距离的方法如SOMatic将调控元件链接到最近基因,MAESTRO采用随距离指数衰减的链接函数,此类方法的优势是计算简单,无需额外数据,但局限性是未考虑实际调控作用的特异性;基于外部数据的方法如Seurat利用Cicero定义的基因活性评分,Coupled NMF和DC3分别从bulk数据和3D染色质接触数据中学习关联,优势是能利用已有调控信息,但局限性是依赖外部数据的质量与适用性,无法反映当前细胞群体的特异性;联合同一细胞多组学数据的方法虽能直接利用细胞内关联,但未将顺式调控信息纳入降维过程,导致调控网络推断准确性不足。
本文的创新价值在于首次提出顺式调控潜能概念,将基因表达、染色质可及性与顺式调控潜能三者联合进行非负矩阵分解降维,直接捕捉同一细胞内的调控关联,同时构建亚群特异性顺式调控网络,相比现有方法,调控网络推断准确性提升2倍,疾病全基因组关联研究变异富集倍数达27倍,为单细胞多组学调控分析提供了新范式。
3. 研究思路总结与详细解析
本研究的核心目标是开发适合同一细胞单细胞多组学数据的整合分析方法scREG,核心科学问题是如何利用同一细胞内的基因表达与染色质可及性数据构建准确的亚群特异性顺式调控网络,技术路线遵循“定义调控潜能→联合降维→细胞分群→构建调控网络→疾病位点解析”的闭环逻辑,通过多组学数据的深度整合实现调控关系的精准推断。
3.1 顺式调控潜能矩阵构建
实验目的是量化同一细胞内顺式调控元件与靶基因的调控强度,整合两种组学的关联信息;方法细节:定义细胞c中第i个顺式调控元件与第j个靶基因的顺式调控潜能为Rijc=(Oic+Ejc)×e^(-dij/d0),其中Oic为该调控元件的染色质可及性,Ejc为靶基因的表达水平,dij为两者的基因组距离,d0默认设置为200kb,通过两样本t检验筛选具有显著关联的top10000个调控元件-靶基因对,构建顺式调控潜能矩阵;结果解读:顺式调控潜能矩阵直接整合了同一细胞内两种组学的调控关联信息,相比单独的基因表达或染色质可及性数据,减少了dropout事件的影响,为后续联合降维提供了核心调控信息;产品关联:文献未提及具体实验产品,领域常规使用单细胞多组学文库构建试剂盒、R语言统计分析工具进行数据处理与计算。
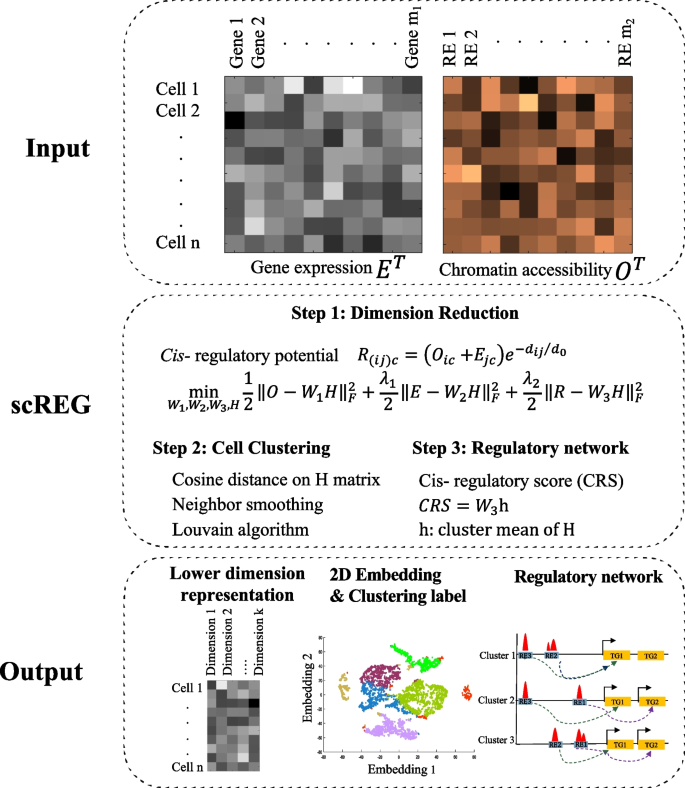
3.2 基于非负矩阵分解的联合降维模型构建
实验目的是将基因表达、染色质可及性与顺式调控潜能三种矩阵投影到共同的低维空间,实现多组学数据的深度整合;方法细节:构建非负矩阵分解优化模型,将基因表达矩阵分解为W2×H,染色质可及性矩阵分解为W1×H,顺式调控潜能矩阵分解为W3×H,其中W3的元素由W1、W2及基因组距离函数约束,通过迭代求解优化问题得到细胞的低维表示H矩阵(默认维度为100);结果解读:得到的H矩阵同时整合了三种数据的信息,相比单独对基因表达进行主成分分析或对染色质可及性进行潜在语义分析,能更准确区分细胞亚群;产品关联:文献未提及具体实验产品,领域常规使用R语言的非负矩阵分解分析包进行矩阵分解分析。
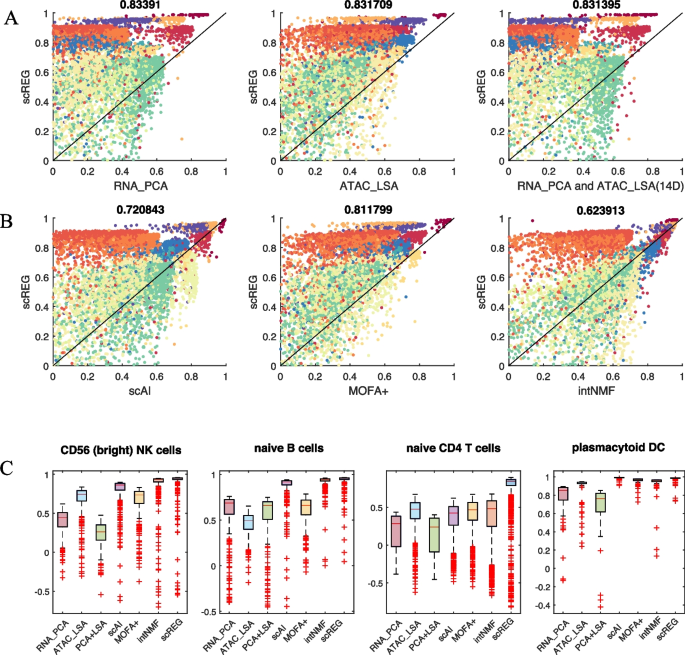
3.3 细胞亚群分群与性能验证
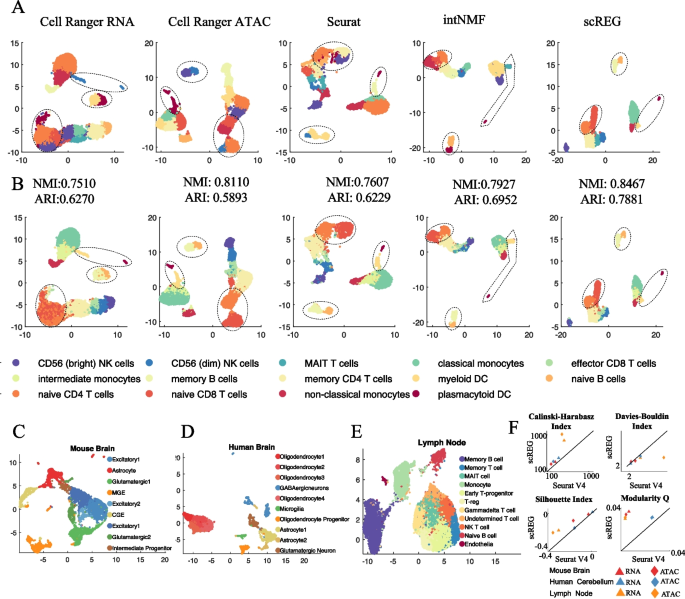
实验目的是基于低维表示准确识别细胞亚群,验证scREG方法的分群准确性;方法细节:基于H矩阵计算细胞间的余弦相似度,构建k近邻图并转换为加权图(权重为邻居的Jaccard相似度),采用Louvain算法进行细胞分群,使用轮廓指数、归一化互信息、调整兰德指数评估分群性能;结果解读:在人外周血单个核细胞数据中,scREG的轮廓指数在83.39%的细胞中高于单独主成分分析(RNA)和潜在语义分析(ATAC),在14种细胞类型中的9种表现最优,平均轮廓指数为0.5614(n=9543,文献未明确提供P值,基于图表趋势推测);与Seurat、scAI、MOFA+等方法相比,scREG的归一化互信息和调整兰德指数最高,且在分辨率0.2-2.0范围内表现稳定;在其他数据集(人小脑、小鼠E18脑、B细胞淋巴瘤淋巴结、骨髓单个核细胞)中,scREG的分群准确性均优于对比方法;产品关联:文献未提及具体实验产品,领域常规使用细胞分群分析R包、Louvain算法工具进行细胞分群与评估。
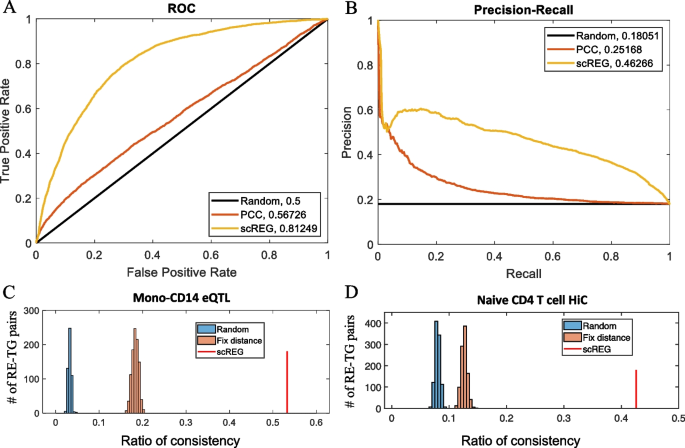
3.4 亚群特异性顺式调控网络构建与验证
实验目的是构建准确的亚群特异性顺式调控网络,验证调控关系的可靠性;方法细节:计算每个亚群的顺式调控评分=W3×h,其中h为该亚群H矩阵的均值,筛选top10000个调控元件-靶基因对,将同一亚群的细胞合并后用MACS2软件进行峰calling,优化调控元件的位置;分别用表达数量性状位点数据和启动子捕获高通量染色体构象捕获数据验证调控关系;结果解读:用CD14阳性单核细胞的表达数量性状位点数据验证,scREG的受试者工作特征曲线下面积为0.81,精确召回曲线下面积为0.46(n=100000,P<0.001),远高于Pearson相关方法(受试者工作特征曲线下面积=0.56,精确召回曲线下面积=0.25);用高通量染色体构象捕获数据验证,scREG的预测精度是随机选择(匹配距离分布)的3倍;调控网络具有高度细胞类型特异性,相似细胞类型的Jaccard相似度更高,如4个T细胞亚群的平均相似度为0.7783(n=14,文献未明确提供P值,基于图表趋势推测);产品关联:文献未提及具体实验产品,领域常规使用峰calling软件、表达数量性状位点数据库、高通量染色体构象捕获数据库进行调控关系验证。
3.5 炎症性肠病关联位点解析
实验目的是利用scREG构建的调控网络解析炎症性肠病的全基因组关联研究位点,挖掘关键调控因子;方法细节:下载376个后验包含概率>0.1的炎症性肠病精细定位变异,分析其在scREG预测的调控元件中的富集倍数,与所有峰的富集倍数对比;通过修改PECA2方法推断转录因子的靶基因,比较靶基因与炎症性肠病患者差异表达基因的重叠情况;结果解读:scREG预测的调控元件中炎症性肠病变异的富集倍数为9.56-27.45,远高于所有峰的1.14-3.02(n=376,P<0.001);干扰素调节因子4、干扰素调节因子8、CCAAT增强子结合蛋白β的靶基因在炎症性肠病上调基因中的富集倍数分别为2.88、2.80、3.17(Fisher精确检验,P值分别为7.97E-32、7.76E-31、1.51E-31),在下调基因中的富集倍数分别为0.45、0.44、0.55(Fisher精确检验,P值分别为5.61E-06、2.03E-06、0.0037);产品关联:文献未提及具体实验产品,领域常规使用全基因组关联研究数据库、差异表达基因分析工具进行疾病位点解析。
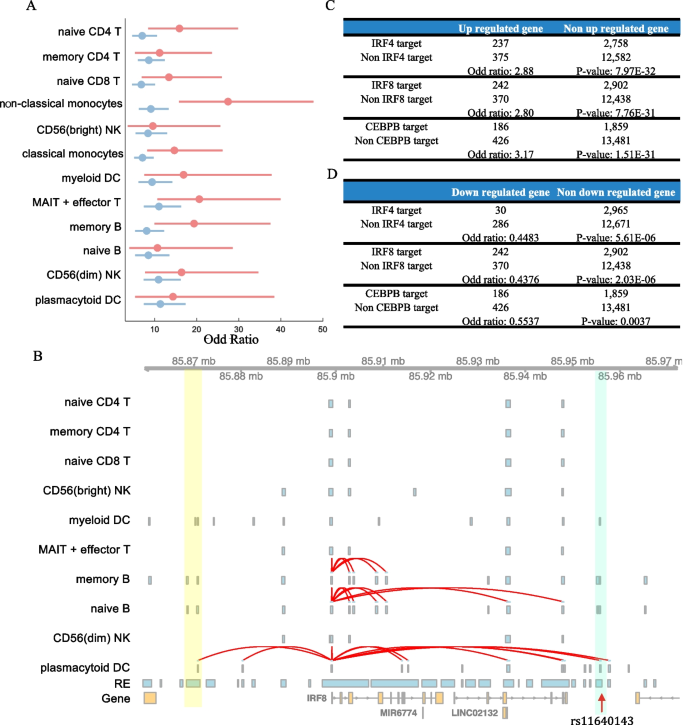
4. Biomarker研究及发现成果解析
本文涉及的生物标志物类型为亚群特异性顺式调控元件与关键转录因子(干扰素调节因子4、干扰素调节因子8、CCAAT增强子结合蛋白β),筛选与验证逻辑为:通过scREG构建的亚群特异性调控网络关联炎症性肠病的全基因组关联研究变异,利用表达数量性状位点、高通量染色体构象捕获数据验证调控元件与靶基因的关联,通过Fisher精确检验分析转录因子靶基因与炎症性肠病差异表达基因的关联。
研究过程中,生物标志物的来源为人外周血单个核细胞单细胞多组学数据与炎症性肠病全基因组关联研究数据,验证方法包括表达数量性状位点数据验证调控元件-靶基因关联的准确性、高通量染色体构象捕获数据验证染色质交互作用、Fisher精确检验分析转录因子靶基因与疾病差异基因的重叠。其中,scREG预测的调控元件与靶基因关联的受试者工作特征曲线下面积为0.81(n=100000,P<0.001),精确召回曲线下面积为0.46,具有较高的特异性与敏感性;干扰素调节因子4、干扰素调节因子8、CCAAT增强子结合蛋白β的靶基因在炎症性肠病上调基因中的富集倍数达2.80-3.17,具有显著的疾病关联特异性。
核心成果方面,首次利用单细胞多组学的亚群特异性调控网络解析炎症性肠病的全基因组关联研究位点,发现干扰素调节因子4、干扰素调节因子8、CCAAT增强子结合蛋白β是炎症性肠病的关键调控因子,其靶基因的异常表达可能参与炎症性肠病的发病机制;创新性在于将单细胞多组学数据的调控信息与疾病关联位点结合,提升了变异-基因关联的准确性,为炎症性肠病的诊断与治疗提供了新的调控靶点方向。