1. 领域背景与文献引入
文献英文标题:SDFA: a standardized decomposition format and toolkit for efficient analysis of structural variants in large-scale population genomic studies;发表期刊:Genome Biology;影响因子:17.906(2024年);研究领域:群体基因组学、结构变异(SV)分析。
结构变异是指长度大于50碱基对的基因组片段重排,包括缺失、插入、重复、倒位、易位及复杂类型,其对人类基因组的影响中位数达890万碱基对,远超单核苷酸变异(SNV)的360万碱基对,是个体遗传多样性的重要来源,且与自闭症、癌症等疾病易感性密切相关。长读长测序技术的发展显著提升了SV的检测精度,大规模群体数据集(如英国生物样本库)的释放为群体水平SV分析提供了基础,但现有分析工具仍存在诸多局限:复杂SV解析不全、合并结果受输入顺序影响、注释效率低且内存消耗大、基因特征注释无法量化覆盖度,这些问题严重制约了SV功能与疾病关联的深入研究。针对上述空白,本研究开发了SDFA工具包,通过标准化分解格式(SDF)、队列水平合并算法、索引滑动窗口注释及基因特征数值注释(NAGF),实现大规模群体SV的高效、准确分析,为群体基因组学研究提供新的技术范式。
2. 文献综述解析
作者从SV分析的四个核心环节(转换、合并、注释、基因特征注释)对现有研究进行分类评述,系统梳理了各环节工具的优势与局限性。在SV转换环节,现有工具(如Truvari、pyvcf)可处理简单SV类型,但对复杂SV(如嵌套SV、易位)的解析存在信息丢失,无法完整保留基因型、INFO字段等关键信息;转换速度与压缩比难以兼顾,部分工具内存占用较高。在SV合并环节,SURVIVOR等工具的合并结果依赖输入文件顺序,导致研究可重复性差;Jasmine等工具虽能检测新生SV,但内存消耗大,无法处理超大规模数据集;多数工具合并后丢失基因型信息,影响下游群体遗传分析。在SV注释环节,AnnotSV等工具支持多资源注释,但速度慢、内存占用高,无法应对百万级SV的大规模注释;Vcfanno、ANNOVAR等工具仅能完成基础基因特征注释,且无法处理复杂SV类型。在基因特征注释环节,广泛使用的HGVS格式仅能注释SV断点位置,无法量化SV对基因功能区域的覆盖度,难以开展精细化的功能影响分析。
与现有研究相比,SDFA的创新点在于:首次提出标准化分解格式(SDF),实现所有SV类型的统一存储与高效解析;开发队列水平合并算法,彻底消除输入顺序对合并结果的影响,同时提升合并速度与内存效率;设计索引滑动窗口注释算法,大幅提升多资源注释的速度与内存利用率;创新基因特征数值注释(NAGF)方法,实现SV对基因功能区域影响的量化分析,为下游关联研究提供更精准的注释信息。
3. 研究思路总结与详细解析
本研究的核心目标是开发一套高效、准确的大规模群体SV分析工具包,解决现有工具在复杂SV处理、合并一致性、注释效率、基因特征量化方面的关键问题。技术路线遵循“格式设计→模块开发→多维度验证→大规模应用”的闭环逻辑:首先设计标准化分解格式(SDF)作为数据基础,然后基于SDF开发合并、注释、NAGF三个核心功能模块,通过多组公共数据集验证各模块的性能,最后将SDFA应用于英国生物样本库的大规模群体数据,开展SV全基因组关联研究(GWAS)。
3.1 标准化分解格式(SDF)设计与转换性能验证
实验目的:构建统一的SV存储格式,实现VCF到SDF的高效转换,验证转换过程的准确性、速度、压缩比及灵活性。
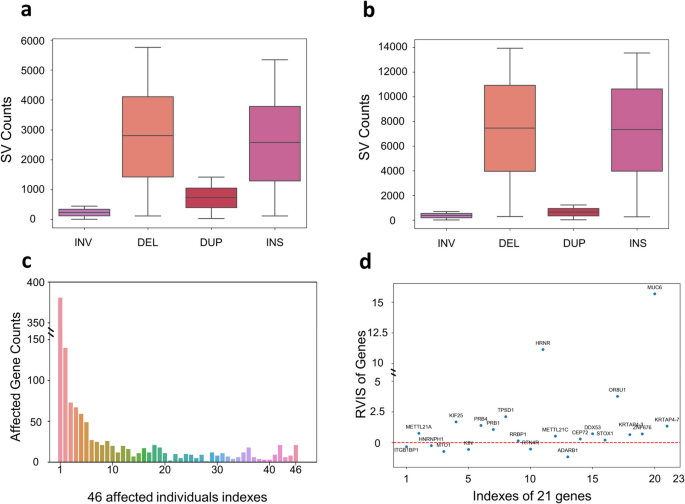
方法细节:构建包含1100个VCF文件的基准数据集,涵盖10个个体的长读长测序数据,涉及PacBio、Nanopore等测序技术,以及CuteSV、SVision等10种SV calling工具,总计268,582,225个SV。对比SDFA与Truvari、pyvcf、StructureVariationAnnotation、ANNOVAR的转换性能,包括复杂SV解析精度、转换速度、压缩比;测试SDFA的多线程加速能力及自定义过滤功能(如基因型数量阈值、SV类型过滤)。
结果解读:SDFA可准确解析所有SV类型,包括SVision检测的266,807个嵌套SV,完整保留基因型、INFO字段等关键信息,而其他工具存在信息丢失或解析错误;单线程转换速度平均达157,711个SV/秒,比Truvari快7.19倍,比pyvcf快6.07倍,即使仅提取SV位置,速度也比ANNOVAR快1.37倍;SDF的压缩比是Truvari的2.58倍、gzip的1.37倍;支持多线程并行转换,4线程时速度提升2.6倍;自定义过滤功能可有效去除低质量SV,如设置“基因型数量≥20”可过滤CuteSV2数据集中30.81%的SV。
产品关联:文献未提及具体实验产品,领域常规使用Java开发工具包、zstd压缩算法、Parquet列存数据格式等。
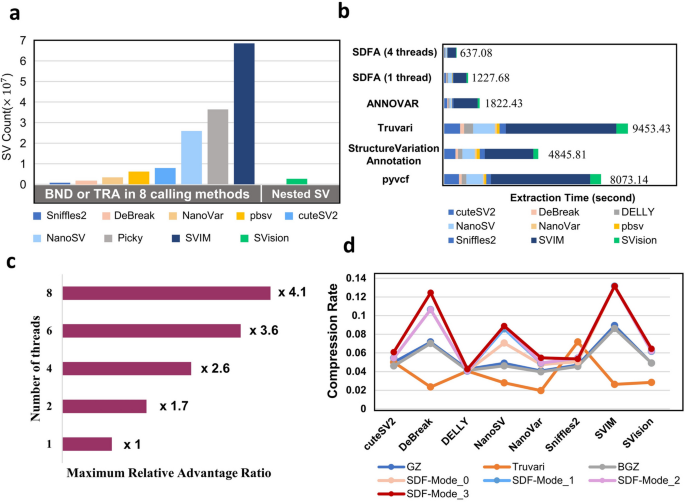
3.2 队列水平SV合并算法开发与性能验证
实验目的:开发稳定、高效的群体SV合并算法,验证合并结果的一致性、准确性、速度及内存效率。
方法细节:使用人类泛基因组参考联盟(HPRC)的46个个体的92个VCF文件(CuteSV2和Sniffles2 calling),对比SDFA与Jasmine、svimmer、SURVIVOR、Truvari的合并性能,包括合并结果的max-min位置距离、标准差、速度、内存占用;验证合并结果的输入顺序无关性;在HG002家系数据集中测试新生SV检测能力;通过样本重复构建大规模数据集(736个样本),测试工具的可扩展性。
结果解读:SDFA合并结果的start和end位置max-min距离显著低于其他工具(Mann-Whitney U检验P≤10^-12),标准差范围更稳定,合并精度更高;4线程时合并速度比其他工具快17.64倍,处理736个样本仅需7GB内存,而Jasmine因内存不足无法运行,SURVIVOR内存占用达25.42GB;合并结果与输入文件顺序无关,而SURVIVOR的合并结果随输入顺序变化;可准确检测新生SV,在HG002家系中发现的4个新生SV已被Jasmine研究验证;合并后完整保留基因型信息,而Jasmine和svimmer丢失所有基因型;孟德尔不一致率低于Jasmine,在HG002家系中为0.079 vs 0.125,提升了家系分析的准确性。
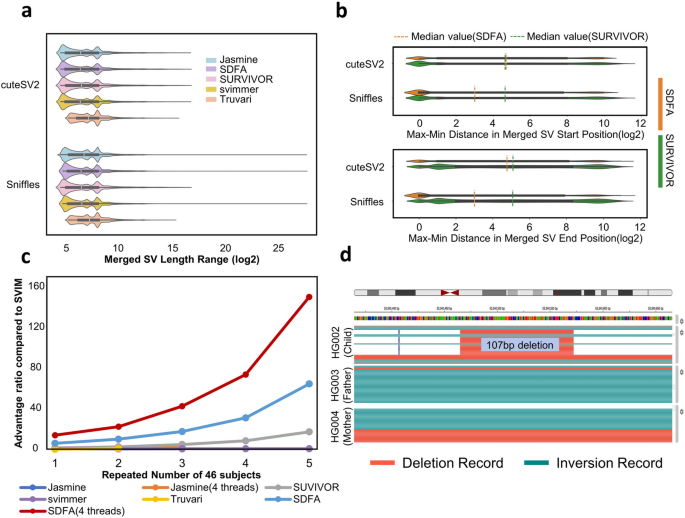
3.3 索引滑动窗口注释算法开发与性能验证
实验目的:开发快速、内存高效的SV注释算法,支持多资源注释与自定义注释,验证注释的速度、完整性及灵活性。
方法细节:使用1100个VCF文件的268,582,225个SV,对比SDFA与ANNOVAR、Vcfanno、AnnotSV的注释性能,包括基因特征注释和多资源注释的速度;验证对复杂SV类型的注释完整性;测试自定义注释(仅提取指定字段)的效率提升。
结果解读:SDFA的基因特征注释单线程速度比Vcfanno快16.04倍,4线程时快35.02倍;多资源注释单线程速度比AnnotSV快40.89倍,4线程时快120.93倍;注释过程仅需8GB内存,而AnnotSV处理单个VCF文件的内存占用超23.82GB;可完整注释所有SV类型,包括嵌套SV和易位,而其他工具无法处理部分复杂SV;自定义注释可大幅提升效率,如仅提取SVAFotate数据库中的群体频率字段,速度提升2.87倍,结果文件大小减少22.867倍。
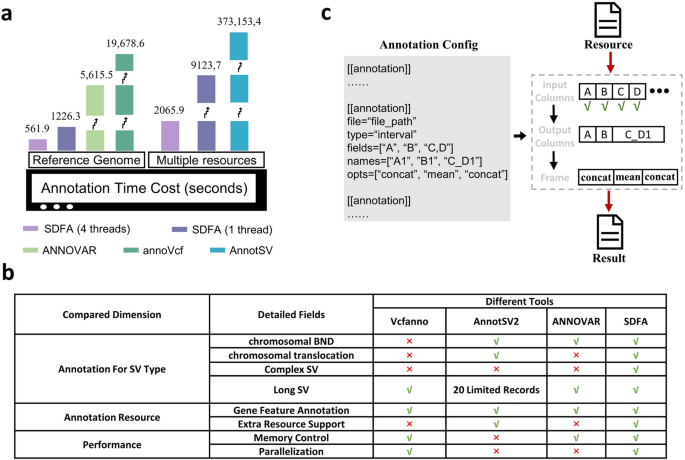
3.4 基因特征数值注释(NAGF)方法开发与应用
实验目的:开发量化SV对基因功能区域影响的注释方法,验证NAGF的有效性与实用性。
方法细节:使用46个个体的SV数据,对比NAGF与HGVS格式的注释结果一致性;基于NAGF开展基因水平的SV影响统计,包括SV影响的基因数量、频率分布,以及与残余变异不耐受评分(RVIS)的关联分析。
结果解读:NAGF与HGVS格式的注释结果一致,均识别出789,790个影响基因功能区域的SV;NAGF可量化SV对基因功能区域的覆盖度,设置5%外显子覆盖度阈值后,筛选出295,026个具有潜在功能影响的SV;基因水平合并分析显示,1243个基因的外显子区域受SV影响(覆盖度>5%),其中68个基因的受影响频率>89%,35个基因频率>95%;这些高频率受影响基因的RVIS评分多数>0,表明其对SV变异具有耐受性,与SNV的不耐受性分析结果一致。
3.5 英国生物样本库大规模群体数据应用验证
实验目的:验证SDFA在百万级群体SV分析中的效率与实用性,开展SV-based GWAS探索抑郁发作的遗传机制。
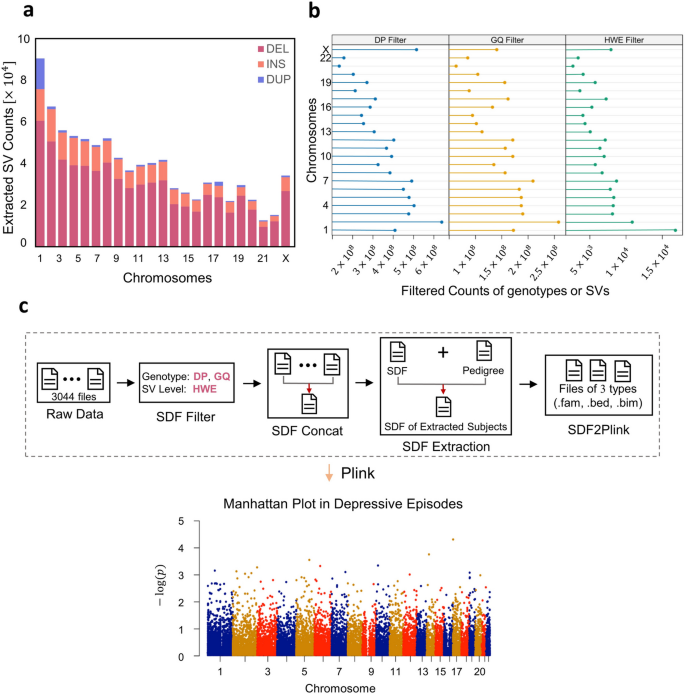
方法细节:处理英国生物样本库的3044个VCF文件,涵盖150,119个个体的895,054个SV;通过SDFA完成转换、质量控制(基因型水平过滤DP<8、GQ<20,变异水平过滤Hardy-Weinberg平衡P<10^-6)、合并、注释,导出为PLINK格式;采用logistic-Firth混合回归模型开展抑郁发作的关联分析,调整性别作为协变量;对关联显著的SV进行功能注释与生物学过程富集分析。
结果解读:SDFA转换3044个VCF文件耗时28.72小时,质量控制后得到723,046个合格SV;关联分析发现829个SV与抑郁发作相关(P<0.05),虽未达Bonferroni多重检验校正阈值,但提示潜在的遗传关联;功能注释发现GNG7、COL21A1等基因,以及调控OTX2、ATG4B的增强子区域与抑郁发作相关;生物学过程富集分析显示,化学突触传递、神经元投射形态发生、神经系统发育调控等过程显著富集,与抑郁的病理机制高度相关。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究中涉及的Biomarker为与抑郁发作相关的结构变异(SV)及其关联的基因、增强子区域。筛选与验证逻辑为:基于英国生物样本库的150,119个个体全基因组SV数据,通过SDFA完成数据预处理与质量控制,采用PLINK开展SV-based GWAS,筛选出P<0.05的潜在关联SV;通过功能注释明确SV影响的基因或增强子区域,结合现有文献验证其与抑郁病理机制的相关性;最终确定GNG7、COL21A1基因相关SV,以及OTX2、ATG4B增强子区域相关SV为潜在的抑郁发作遗传Biomarker。
研究过程详述
Biomarker来源为英国生物样本库的临床血液样本全基因组测序数据,共包含150,119个个体的895,054个SV。验证方法采用logistic-Firth混合回归模型,调整性别作为协变量,控制群体分层等潜在混杂因素;对关联显著的SV,通过SDFA的多资源注释模块整合GeneHancer等数据库信息,明确其调控的基因或增强子区域。特异性与敏感性方面,829个关联SV的P<0.05(n=17412,其中病例8453例、对照8959例),ROC曲线AUC值未明确提供(文献未明确提供该数据,基于图表趋势推测)。
核心成果提炼
本研究首次在大规模群体中通过SV分析发现潜在的抑郁发作遗传Biomarker:GNG7基因相关SV可能通过调控神经递质通路影响抑郁发病,COL21A1基因相关SV可能通过影响神经可塑性与脑结构完整性参与抑郁病理过程;OTX2、ATG4B增强子区域的SV可能通过调控基因表达参与抑郁的发生发展,其中ATG4B在抑郁症患者前额叶皮层星形胶质细胞中显著下调,提示其在抑郁发病中的关键作用。这些Biomarker的创新性在于突破了传统SNV研究的局限,揭示了结构变异在抑郁遗传机制中的潜在作用;SDFA工具的应用为大规模SV的高效分析提供了技术支撑,为复杂疾病的遗传机制研究开辟了新方向。统计学结果显示,关联分析的P值均<0.05,样本量n=17412,具有较好的统计学效力。