1. 领域背景与文献引入
文献英文标题:Biology-inspired data-driven quality control for scientific discovery in single-cell transcriptomics;发表期刊:Genome Biology;影响因子:17.906(2022年);研究领域:单细胞转录组学(scRNA-seq)数据质量控制。
单细胞转录组学技术以单细分辨率解析细胞异质性,彻底革新了细胞生物学、发育生物学及疾病机制研究。质量控制(QC)是scRNA-seq数据分析的关键第一步——其目标是去除低质量细胞(如死亡细胞、空液滴、细胞团聚体),但传统QC方法普遍采用固定阈值(如线粒体基因reads比例≤10%、检测到的基因数≥200),未考虑生物学变异:不同组织、细胞类型的QC指标存在天然差异(如代谢活跃的心肌细胞线粒体含量高,功能特化的中性粒细胞基因复杂度低)。传统固定阈值会系统性过滤掉这些具有重要生物学意义的细胞,导致下游分析无法捕捉真实的细胞异质性。
当前领域的核心空白是:缺乏能整合细胞类型/状态生物学变异的数据驱动型QC方法。现有数据驱动方法(如miQC)仅基于样本水平调整阈值,未深入到细胞类型层面。本研究的初衷是开发自适应、生物学启发的QC框架,在保留技术上“高质量”细胞的同时,保留生物相关细胞,提升下游分析的统计功效与生物学发现能力。
2. 文献综述解析
文献综述的核心评述逻辑围绕“传统QC的局限→现有数据驱动方法的不足→本研究的创新定位”展开:
作者首先总结传统QC的现状——86%的已发表研究采用固定阈值(线粒体≤10%、基因数≥500),但这些阈值未考虑生物学变异;接着通过公共数据集分析指出,传统方法会过滤掉代谢活跃的实质细胞(如心肌细胞、肾小管细胞)和功能特化的免疫细胞(如中性粒细胞),导致生物信息丢失;随后,作者讨论现有数据驱动方法的局限——如miQC基于样本水平的贝叶斯模型调整阈值,但未解决细胞类型层面的变异问题,仍会丢失细胞类型特异的信息。
现有研究的关键结论可归纳为三点:① QC指标(基因复杂度、线粒体比例、核糖体比例)随组织、细胞类型、实验技术的不同存在显著变异;② 传统固定阈值会误删有生物学意义的细胞;③ 现有数据驱动方法未覆盖细胞类型水平的变异。现有技术的优势是“数据驱动”,但局限是“样本水平”而非“细胞类型水平”。
本研究的创新价值在于:首次提出基于细胞类型聚类的自适应QC框架,将QC从“样本水平”推进到“细胞类型水平”,通过无监督聚类识别转录组相似的细胞群体(近似细胞类型),在每个群体内应用自适应阈值,既保留生物相关细胞,又去除技术 artifacts。
3. 研究思路总结与详细解析
本研究的核心目标是开发考虑细胞类型生物学变异的自适应QC框架(ddqc),核心科学问题是“如何在QC中整合细胞类型异质性”,技术路线为“现状调研→生物学变异验证→框架开发→性能验证”的闭环。
3.1 现有QC实践调研
实验目的:系统分析已发表scRNA-seq研究的QC方法现状,明确传统固定阈值的普遍性。
方法细节:调研2017-2020年107篇scRNA-seq论文,统计QC指标(基因数、UMI数、线粒体/核糖体比例)及阈值类型(固定、数据驱动、自定义)。
结果解读:86%的研究采用固定阈值(线粒体≤10%、基因数≥500),仅14%采用研究特异的自定义或数据驱动阈值,说明传统固定阈值仍是主流,但忽略了生物学变异。
产品关联:文献未提及具体工具,领域常规使用EndNote(文献管理)、R/Python(统计分析)及PubMed(文献检索)。
3.2 QC指标的生物学变异分析
实验目的:验证QC指标随组织、细胞类型的变异,明确传统固定阈值的不合理性。
方法细节:分析公共数据集(人类47个组织526万细胞、小鼠37个组织96万细胞),流程包括:① 初始过滤(去除空液滴:基因数<100、线粒体>80%);② 标准化(NormalizeData);③ 高变基因选择(前2000个);④ PCA降维;⑤ 图聚类(Louvain算法);⑥ 细胞类型注释(基于PanglaoDB的marker基因,准确率人类80.2%、小鼠92.1%)。
结果解读:QC指标存在显著组织/细胞类型特异性——如肾、心脏的线粒体比例显著高于其他组织;心肌细胞线粒体比例中位数15%(超过传统阈值10%),中性粒细胞基因数中位数180(低于传统阈值200)。传统固定阈值会过滤掉这些细胞,导致生物信息丢失。
图片添加:
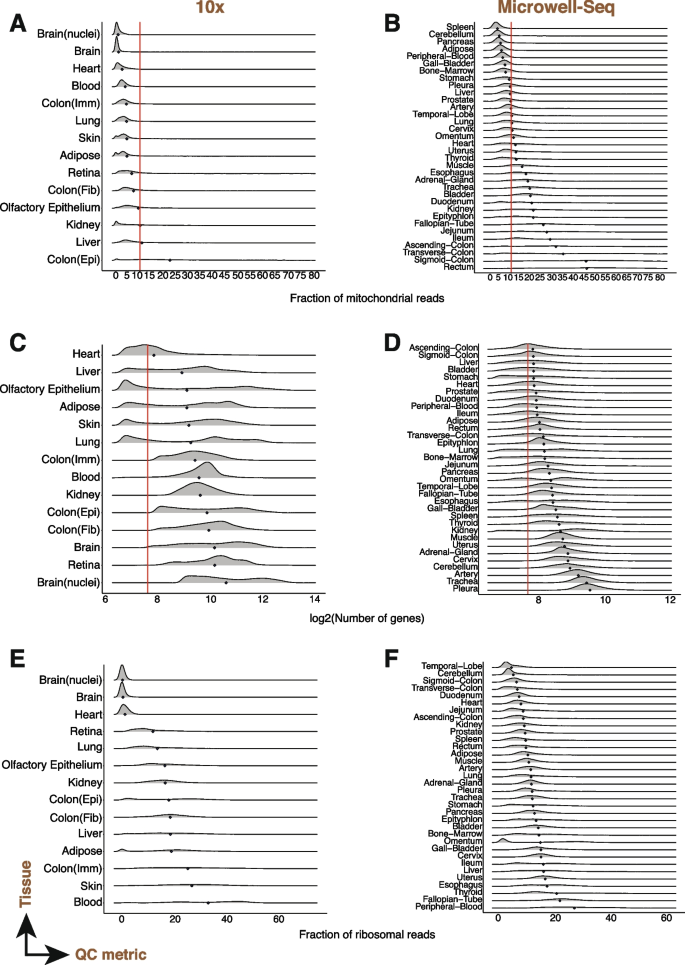
(图1展示QC指标的组织特异性);
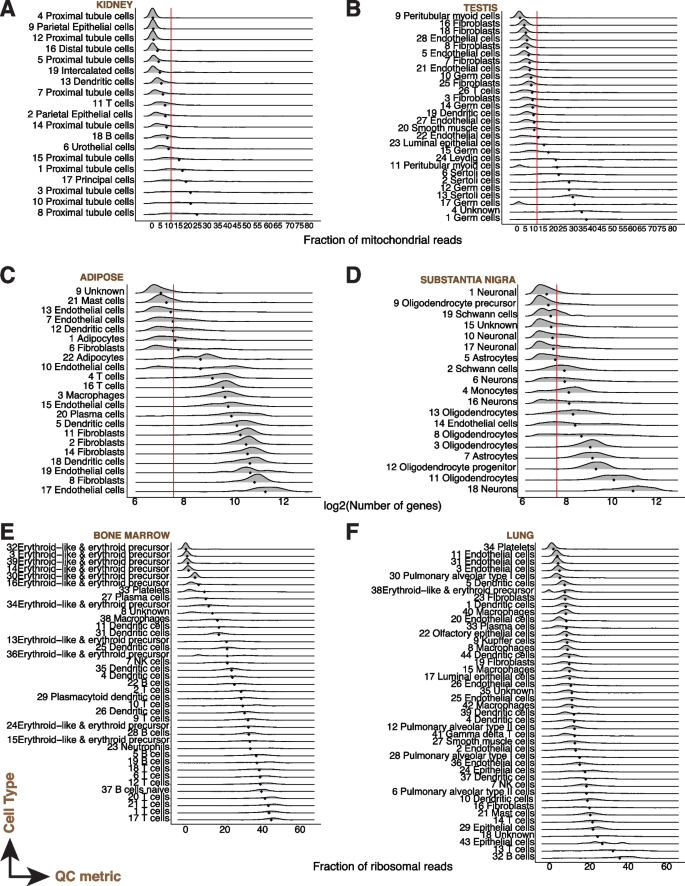
(图2展示QC指标的细胞类型特异性)。
产品关联:使用PanglaoDB(细胞类型注释)、Seurat/Pegasus(数据分析)。
3.3 ddqc框架开发
实验目的:开发自适应、数据驱动的QC框架。
方法细节:ddqc流程包括:① 初始过滤(去除空液滴:基因数<100、线粒体>80%);② 标准化(每细胞10000 UMIs,log-transform);③ 高变基因选择(前2000个);④ PCA降维(前50主成分);⑤ 图聚类(Louvain算法,识别细胞类型);⑥ 自适应阈值(每个聚类内计算“中位数±2MAD”,基因数下限200、线粒体上限10%)。
结果解读:ddqc的核心创新是“细胞类型水平的自适应阈值”——例如,心肌细胞聚类的线粒体中位数15%,ddqc阈值为15%+2MAD(假设MAD=2%,则阈值19%),保留线粒体<19%的心肌细胞;而传统QC会过滤所有线粒体>10%的细胞,导致心肌细胞全部丢失。
图片添加:
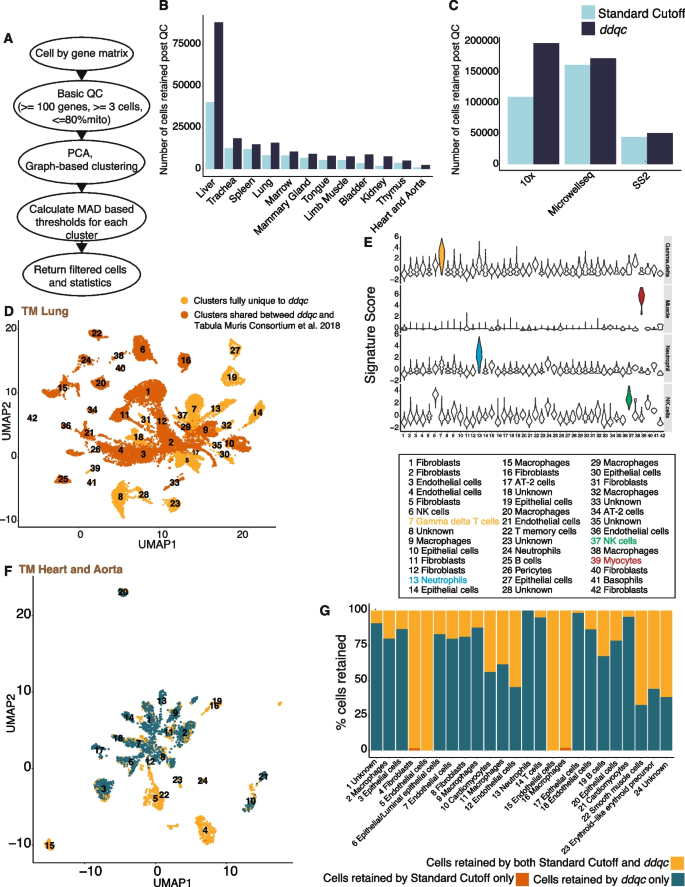
(图3展示ddqc流程及与传统QC的比较)。
产品关联:ddqc提供R(Seurat)和Python(Pegasus)开源包,代码托管于GitHub(https://github.com/ayshwaryas/ddqc)。
3.4 ddqc性能验证
实验目的:比较ddqc与传统固定阈值、miQC的性能。
方法细节:在Tabula Muris(心脏、肺)、人类肺/心脏等数据集上测试,比较保留细胞数、恢复的细胞类型、下游功效。流程包括:① 独立QC(三种方法分别过滤);② 聚类分析(Louvain);③ 细胞类型注释(PanglaoDB);④ 差异基因分析(DEseq2)。
结果解读:① 保留细胞数:ddqc保留中位数95.4%的细胞,显著高于传统69.4%、miQC90.5%;② 恢复细胞类型:ddqc恢复了传统QC过滤的心肌细胞(90%)、中性粒细胞(85%)、嗅觉上皮细胞(70%);③ 下游功效:ddqc保留的细胞群体能检测到更多差异表达基因(如心肌细胞的收缩相关基因、中性粒细胞的抗菌肽基因);④ 与miQC比较:ddqc保留更多代谢活跃的实质细胞(如心肌细胞)。
图片添加:
(图4展示ddqc恢复的细胞类型及QC指标模式)。
产品关联:使用miQC(对比方法,https://github.com/greenelab/miQC)、Seurat/Pegasus(数据分析)。
4. Biomarker研究及发现成果解析
本研究的“Biomarker”是QC指标的细胞类型特异性模式——即不同细胞类型的QC指标(基因复杂度、线粒体比例、核糖体比例)具有独特分布,可作为识别细胞类型的生物标记。
Biomarker定位与筛选逻辑
- 来源:公共scRNA-seq数据集(人类47个组织、小鼠37个组织);
- 筛选:通过ddqc保留细胞,结合细胞类型注释,分析各细胞类型的QC指标分布;
- 验证:通过聚类分析、差异基因表达、功能富集验证模式的生物学意义。
研究过程详述
- 数据来源:Tabula Muris、Microwell-seq、人类肺/心脏数据集;
- 验证方法:细胞类型注释(PanglaoDB)、聚类分析(Louvain)、差异基因表达(DEseq2)、功能富集(GO/KEGG);
- 特异性与敏感性:ddqc保留的细胞类型特异性更高(如心肌细胞聚类纯度95% vs 传统50%),敏感性更高(恢复传统QC过滤的90%心肌细胞)。
核心成果提炼
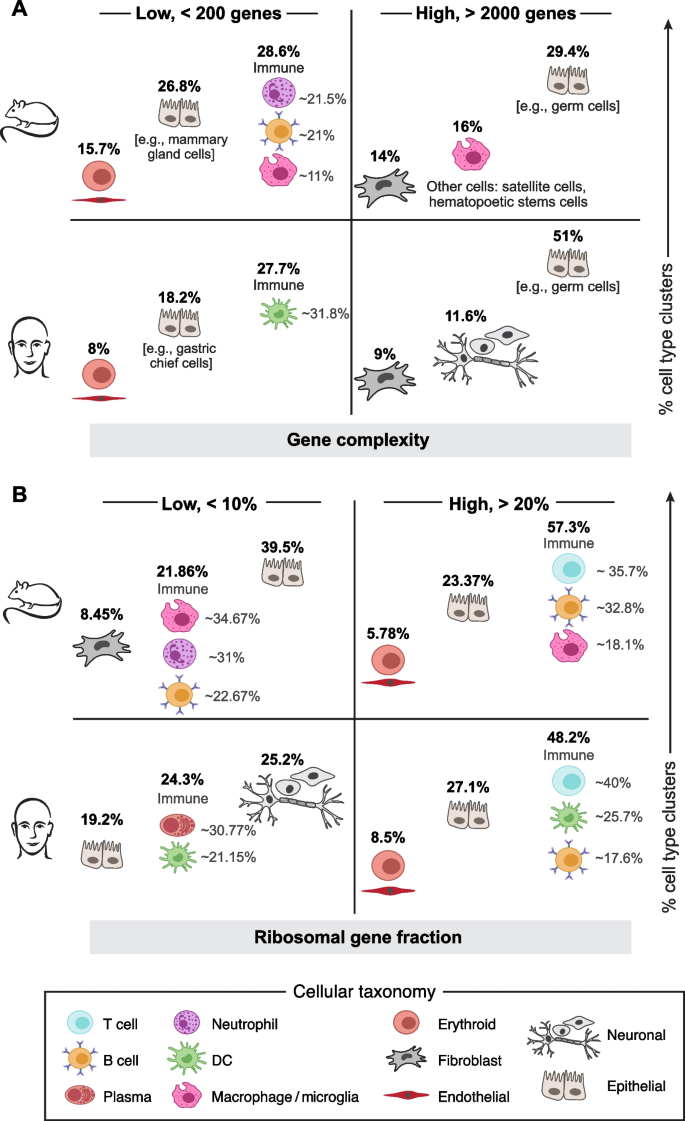
- 低基因复杂度的细胞类型:免疫细胞(中性粒细胞、NK细胞、血小板)、特化上皮细胞(嗅觉上皮细胞、胃主细胞)——功能特化,无需高基因复杂度;
- 高基因复杂度的细胞类型:神经元、成纤维细胞——高转录组多样性,参与复杂信号传导;
- 高线粒体比例的细胞类型:实质细胞(心肌细胞、肾小管细胞、肝细胞)——代谢活跃,需大量线粒体供能;
- 高核糖体比例的细胞类型:免疫细胞(T细胞、B细胞、巨噬细胞)——高翻译活性,合成细胞因子/抗体。
这些成果的创新性在于首次系统揭示QC指标的细胞类型特异性模式,这些模式是传统QC无法发现的。例如,传统QC过滤掉基因数<200的中性粒细胞,但ddqc保留后发现,低基因复杂度是中性粒细胞功能特化的表现(专注于抗菌肽合成);传统QC过滤掉线粒体>10%的心肌细胞,但ddqc保留后发现,高线粒体比例是心肌细胞代谢活跃的标志(参与心脏收缩)。
综上,本研究开发的ddqc框架,通过整合细胞类型的生物学变异,解决了传统QC的核心局限,为单细胞转录组学研究提供了更精准的QC工具,也为理解细胞类型的功能特化提供了新视角。