1. 领域背景与文献引入
文献英文标题:Genetic basis of transcriptome differences between the founder strains of the rat HXB/BXH recombinant inbred panel;发表期刊:Genome Biology;影响因子:未公开;研究领域:分子遗传学(大鼠重组近交系基因组与转录组关联)
下一代测序技术兴起后,基因组变异检测实现规模化,但变异对基因功能的调控机制仍不明确,重组近交系(RI)是研究遗传变异对分子表型(如基因表达)顺式和反式作用的有力工具。领域发展关键节点包括2005年重组近交系首次用于表达数量性状位点(eQTL)的大规模鉴定,2010年后全基因组重测序技术开始系统应用于重组近交系创始品系的变异解析。当前研究热点聚焦于不同类型基因组变异与转录组的关联机制、功能变异的筛选策略,未解决的核心问题是缺乏对结构变异等复杂变异类型调控转录组的系统性解析,尤其是大鼠模型中此类研究仍存在空白。
本研究针对上述领域空白,以大鼠HXB/BXH重组近交系的两个创始品系——自发性高血压大鼠(SHR/OlaIpcv)和褐挪威大鼠衍生系(BN-Lx/Cub)为研究模型,通过全基因组重测序与转录组测序的整合分析,系统性鉴定基因组变异图谱并解析其与转录组差异的关联机制,为遗传变异的功能注释提供了重要的模型支持和数据资源。
2. 文献综述解析
作者对领域内现有研究的分类维度主要涵盖基因组变异类型(单核苷酸变异、插入缺失变异、拷贝数变异/结构变异)和研究模型(人类群体、小鼠重组近交系、大鼠重组近交系)两个层面,系统梳理了不同模型下遗传变异与转录组关联研究的进展与局限性。
现有研究的关键结论显示,人类群体中基因组变异数量庞大,但由于遗传异质性强,难以精准区分功能变异与中性变异;小鼠重组近交系研究表明结构变异对表型的直接影响相对较小,但强数量性状位点(QTL)区域显著富集结构变异;大鼠重组近交系此前已成功用于表达数量性状位点的鉴定和转录调控网络解析,但缺乏对创始品系全基因组变异与转录组差异的系统性整合分析。技术方法层面,重组近交系模型的优势在于遗传背景均一,可重复验证,有效降低实验误差;下一代测序技术可实现全基因组范围内的变异规模化检测,但现有研究对结构变异的解析深度不足,且缺乏对不同变异类型调控转录组的对比分析。
本研究的创新价值在于首次全面构建了大鼠HXB/BXH重组近交系创始品系的全基因组变异图谱,涵盖单核苷酸变异、插入缺失变异、拷贝数变异和结构变异四大类,并通过转录组测序数据的整合分析,系统性对比了不同变异类型对转录组差异的调控作用,尤其是揭示了基因重复等结构变异与转录组差异的强关联,填补了大鼠重组近交系遗传-转录关联研究的空白,为后续功能变异的筛选和验证提供了重要基础。
3. 研究思路总结与详细解析
本研究的整体框架以“全基因组变异解析-转录组差异检测-关联机制解析-关键变异验证”为核心逻辑,研究目标是全面鉴定大鼠HXB/BXH重组近交系创始品系的基因组变异,解析不同类型变异对转录组差异的调控机制;核心科学问题聚焦于结构变异等复杂变异如何调控基因表达和转录本结构;技术路线形成了从基因组到转录组的完整闭环,确保研究结论的严谨性和可靠性。
3.1 全基因组重测序与基因组变异检测
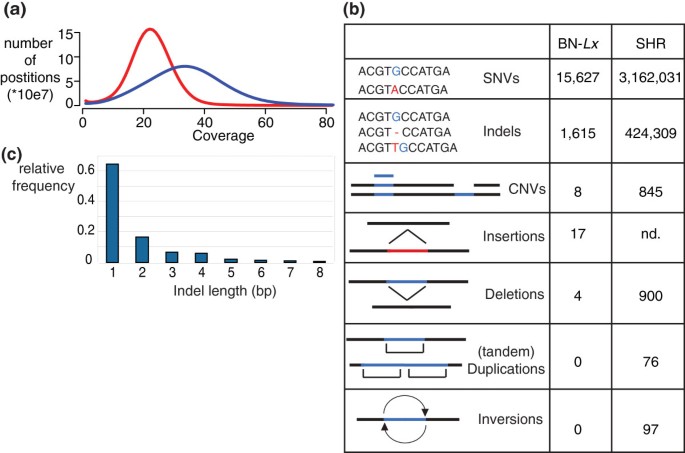
实验目的为全面鉴定SHR和BN-Lx两个创始品系的各类基因组变异,包括单核苷酸变异、插入缺失变异、拷贝数变异和结构变异,为后续关联分析提供基础数据。方法细节上,研究团队对SHR基因组进行了深度测序,将覆盖度从之前的10×提升至23×,同时对BN-Lx基因组进行了33×覆盖度的测序,结合两种独立的变异检测算法(Samtools+自定义脚本、Genome Analysis Toolkit)进行变异识别,并利用BN参考品系(Eve)的32×覆盖度测序数据过滤参考基因组误差;采用动态窗口覆盖度分析(DWAC-Seq)和长 mate-pair文库分析检测拷贝数变异和结构变异。结果解读显示,共检测到3177658个单核苷酸变异,其中SHR特有3162031个,BN-Lx特有15627个;425924个插入缺失变异,SHR特有424309个,BN-Lx特有1615个;此外还鉴定出907个拷贝数变异和1094个结构变异。通过毛细管测序验证,单核苷酸变异和插入缺失变异的验证率均为100%(n=70和n=57),证明变异检测结果的准确性。
文献未提及具体实验产品,领域常规使用Illumina HiSeq、AB/Solid等测序平台,QIAquick纯化试剂盒、End-It末端修复试剂盒等文库构建试剂。
3.2 肝脏组织转录组差异分析
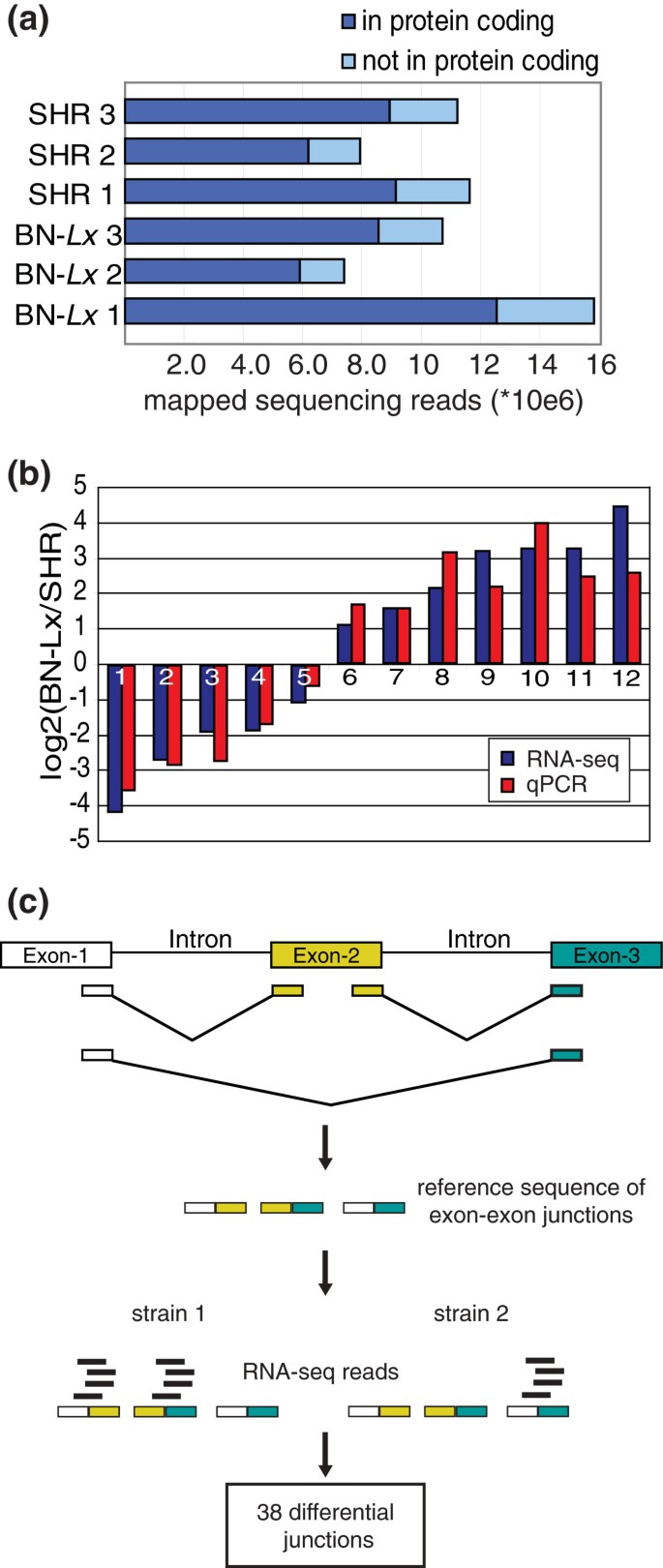
实验目的是检测SHR和BN-Lx两个品系肝脏组织的转录组差异,包括基因表达水平的定量差异和转录本结构的定性差异。方法细节上,研究选取每个品系3只大鼠的肝脏组织,提取总RNA后通过PolyA富集和5"-cap筛选获得全长mRNA,构建链特异性转录组测序文库并进行测序,每样本获得超过740万可比对的测序reads;以映射到基因编码区的归一化reads数作为基因表达量的衡量指标,采用t检验结合样本置换法计算错误发现率(FDR),筛选差异表达基因;通过构建包含所有注释外显子组合的参考转录组,分析外显子-外显子连接的覆盖度差异,鉴定可变剪接事件。结果解读显示,共鉴定出532个差异表达基因,满足表达差异≥2倍且错误发现率<5%的标准;随机选取12个差异表达基因进行定量PCR验证,所有基因的表达差异均显著(P值范围为9.8×10^-9至0.04,n=3),验证了转录组测序结果的可靠性;同时发现36个基因存在可变剪接差异,其中27个为注释的外显子连接,11个为新的外显子组合。
文献未提及具体实验产品,领域常规使用Micro PolyA Purist试剂盒富集mRNA,Promega反转录酶、BioRad MyIQ定量PCR仪等验证试剂与仪器。
3.3 基因组变异与转录组关联分析
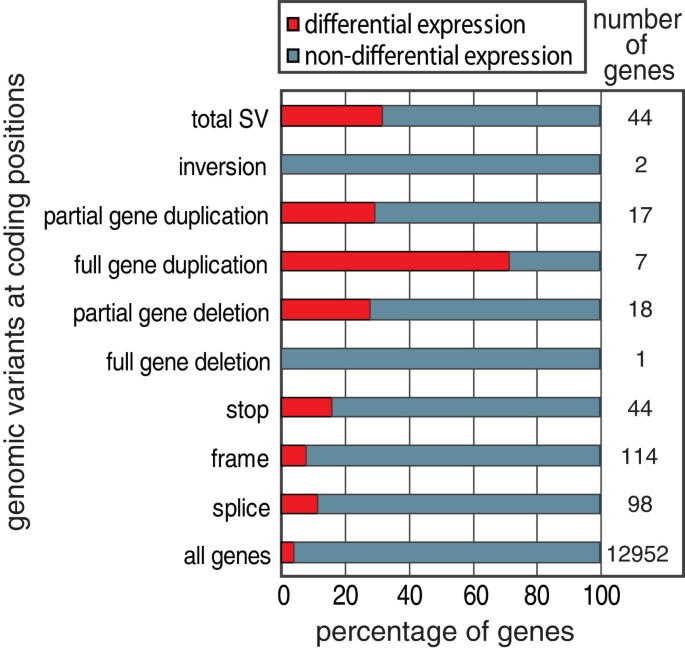
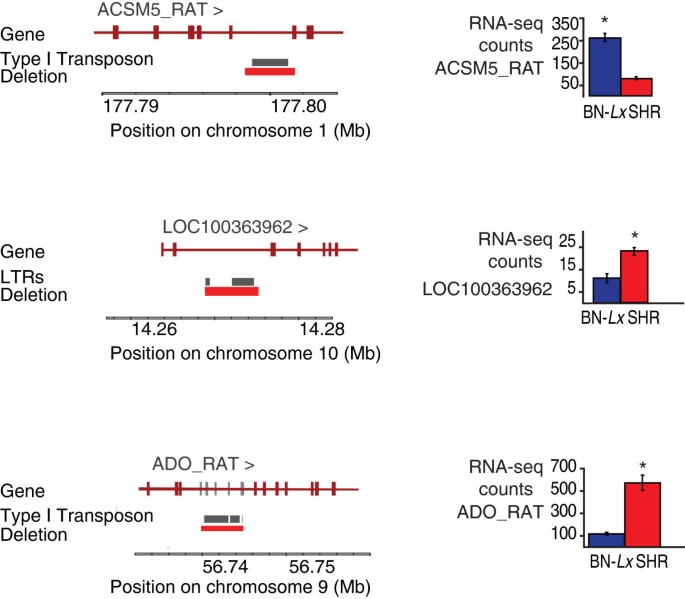
实验目的是解析不同类型基因组变异对转录组差异的调控作用,明确具有功能效应的变异类型及其作用机制。方法细节上,研究团队将单核苷酸变异、插入缺失变异、结构变异分别与差异表达基因、可变剪接基因进行关联分析,计算各类变异所关联的基因中差异表达基因的比例;对关键变异类型(如内含子重复元件缺失、剪接位点变异)进行可视化分析,结合保守性评分判断变异的功能潜力。结果解读显示,结构变异尤其是基因重复与转录组差异的关联最强,7个完全重复且在肝脏中表达的基因中有5个为差异表达基因;终止密码子变异对转录组的影响相对较小,仅4%的差异表达基因包含此类变异,进一步分析发现部分预测的终止密码子变异位于肝脏中不表达的外显子区域,或因基因注释不准确导致预测误差;内含子中重复元件的缺失可能调控基因表达,研究发现4个含内含子长散在元件或长末端重复元件缺失的基因出现差异表达;可变剪接差异与剪接位点变异的关联较弱,仅1个可变剪接事件(Slc22A18基因的外显子跳跃)与剪接位点单核苷酸变异直接相关。
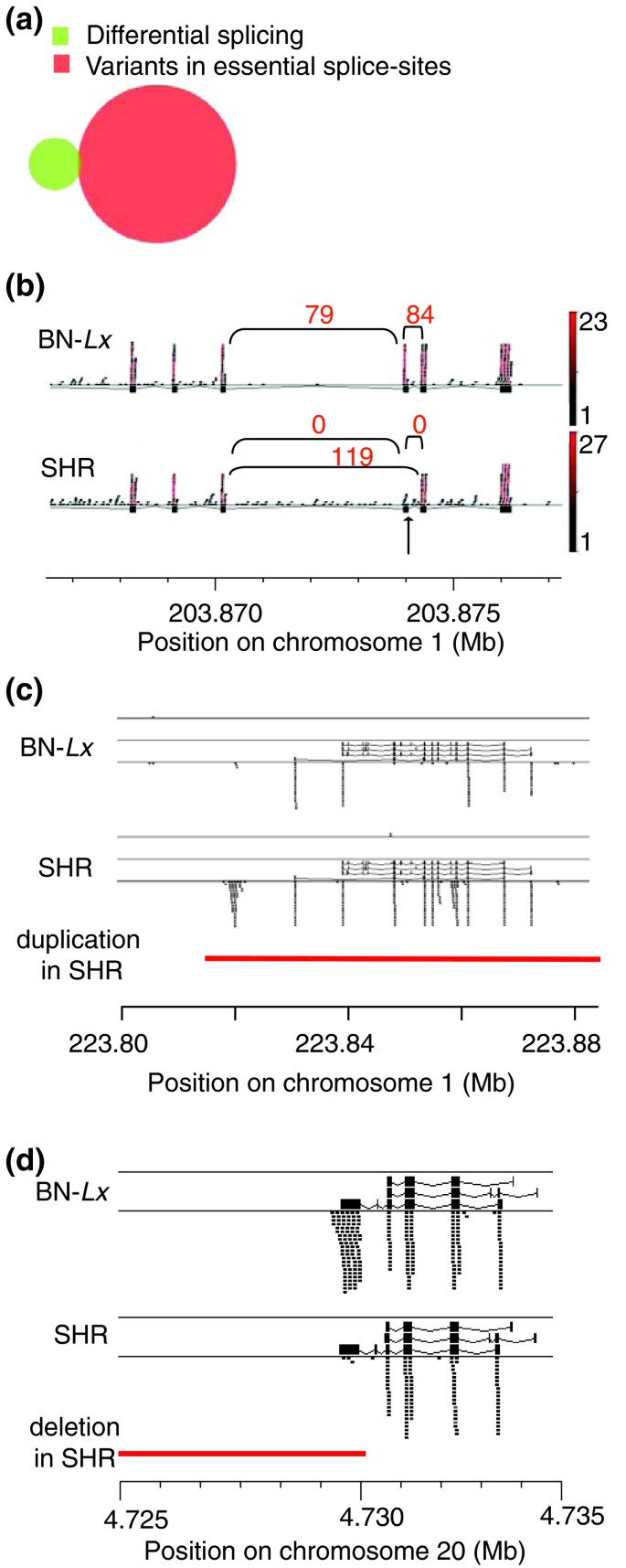
文献未提及具体实验产品,领域常规使用Variant Effect Predictor进行变异功能注释,UCSC基因组浏览器进行基因组数据的可视化分析。
3.4 关键结构变异与转录本功能验证
实验目的是验证基因重复等结构变异对基因表达的调控机制,明确重复基因的转录本来源及表达水平变化。方法细节上,研究团队对Mx2和Layn两个重复基因的基因组区域进行覆盖度分析,通过转录组测序reads的等位基因频率分析,判断转录本是否来自原基因座和重复基因座;同时对比两个品系中基因的表达水平差异。结果解读显示,Mx2和Layn基因在SHR中均存在基因重复变异,其基因组区域的测序覆盖度显著高于BN-Lx;转录组测序数据显示,两个基因的杂合单核苷酸变异位点均检测到两种等位基因的表达,证明转录本同时来自原基因座和重复基因座;且两个基因在SHR中的表达水平均显著上调(n=3,错误发现率<5%),提示基因重复通过增加基因拷贝数或调控元件剂量提升基因表达。
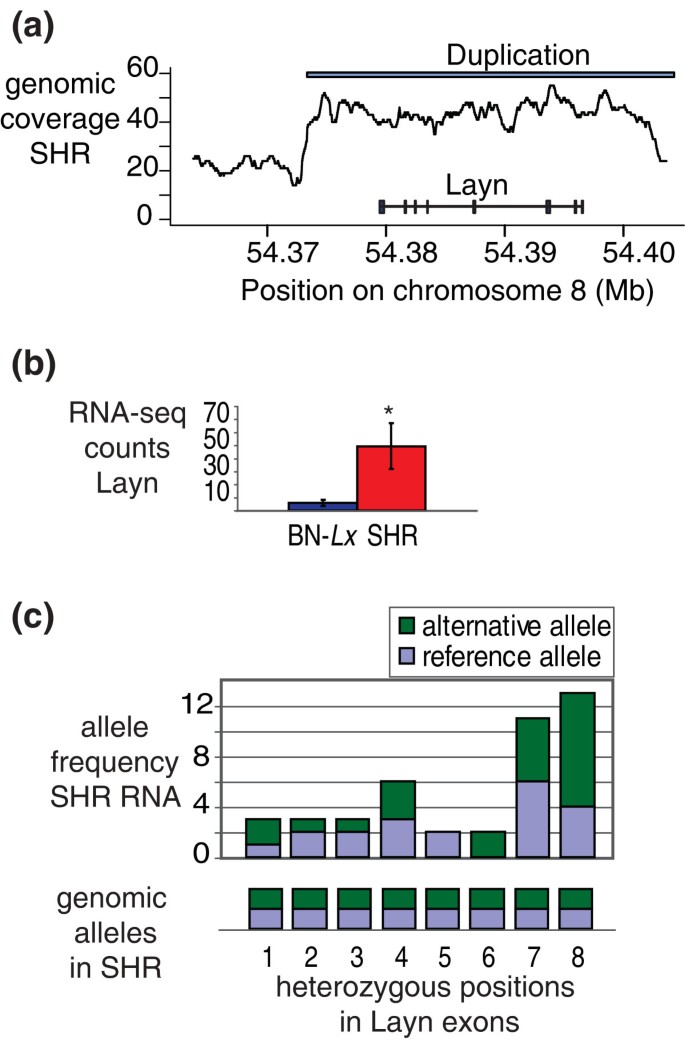
文献未提及具体实验产品,领域常规使用Integrative Genomics Viewer进行测序数据的可视化验证。
4. Biomarker研究及发现成果解析
本研究中涉及的生物标志物为与转录组差异显著关联的基因组变异,涵盖单核苷酸变异、插入缺失变异和结构变异三大类,其中结构变异尤其是基因重复是核心的功能性生物标志物,其筛选与验证逻辑为“全基因组变异检测-转录组差异分析-关联分析筛选候选变异-测序与定量PCR验证”,形成了完整的研究链条。
生物标志物的来源为SHR和BN-Lx两个创始品系的全基因组序列及肝脏组织转录组数据,验证方法包括毛细管测序验证单核苷酸变异和插入缺失变异的真实性,定量PCR验证基因表达差异,转录组测序reads的等位基因分析验证重复基因的转录本来源。特异性与敏感性数据显示,基因重复等结构变异对差异表达基因的预测特异性最高,7个完全重复且表达的基因中有5个为差异表达基因(敏感性71.4%);单核苷酸变异的预测敏感性较低,仅4%的差异表达基因包含终止密码子单核苷酸变异,提示此类变异对转录组的直接调控作用有限。
核心成果提炼显示,本研究首次在大鼠重组近交系中发现基因重复等结构变异是转录组差异的关键调控因子,Mx2和Layn基因的重复变异可显著提升基因表达水平(n=3,错误发现率<5%);创新性在于系统性对比了不同类型基因组变异对转录组的调控作用,揭示了内含子重复元件缺失对基因表达的潜在调控机制;此外,研究还发现部分可变剪接差异可能由非剪接位点的调控区域变异引起,但具体机制仍需进一步验证。这些成果为遗传变异的功能注释提供了重要参考,也为大鼠高血压等疾病模型的分子机制研究提供了新的线索。