1. 领域背景与文献引入
文献英文标题:Subtractive assembly for comparative metagenomics, and its application to type 2 diabetes metagenomes;发表期刊:Genome Biology;影响因子:11.31(2015年);研究领域:比较宏基因组学、2型糖尿病肠道微生物组
宏基因组学通过直接测序环境中全部微生物的基因组DNA,突破了传统纯培养技术的限制,为解析微生物群落组成与功能提供了核心技术支撑。领域共识:2005年比较宏基因组学概念提出后,该领域快速发展,已广泛应用于环境微生物、人类疾病相关微生物组研究,尤其是2012年人类微生物组计划完成后,肠道微生物组与代谢疾病的关联成为研究热点。然而,宏基因组数据具有规模大、物种组成复杂、测序深度不均的特点,传统组装方法在处理宏基因组样本时,难以区分近缘菌株的序列差异,导致组装contig碎片化严重、嵌合序列比例高,极大限制了差异微生物区域的解析效率。当前比较宏基因组学研究的核心未解决问题是,如何高效识别并组装两组宏基因组间的差异序列,同时降低计算资源消耗。针对这一研究空白,本研究提出减法组装策略,通过预先提取差异reads,排除非差异序列的干扰,为大规模比较宏基因组学研究提供新的技术方案,其学术价值在于弥补现有方法在差异区域组装上的不足,提升疾病相关微生物组特征的解析深度。
2. 文献综述解析
作者系统梳理了比较宏基因组学的三类核心研究方法:基于短读长的微生物多样性估计、基于参考数据库的功能注释、基于k-mer序列特征的样本比较。现有研究中,基于短读长的方法(如MetaCluster)能快速估计样本的物种组成,但无法获得完整的基因组或基因序列;基于参考数据库的注释方法(如MEGAN、IMG/M)能实现功能解析,但依赖已知数据库,存在注释偏差;基于k-mer的方法(如Compareads、d₂ˢ dissimilarity)能在无参考基因组的情况下比较样本差异,但未针对差异序列的组装进行优化。传统宏基因组组装方法的局限性在于,直接组装所有reads会引入大量非差异序列,导致计算资源消耗大,且近缘菌株的同源序列会干扰组装过程,产生大量嵌合contig(如Mende等的研究显示,400基因组群落的模拟数据组装后,37%的contig为嵌合序列)。本研究的创新价值在于,首次将减法策略应用于宏基因组组装,通过预先提取差异reads,既降低了计算复杂度,又提升了差异基因组的组装质量,同时无需依赖参考基因组,弥补了现有方法在差异区域组装上的不足,为比较宏基因组学提供了一种互补性的技术方案。
3. 研究思路总结与详细解析
本研究的核心目标是开发并验证一种适用于比较宏基因组学的减法组装方法,核心科学问题是如何高效识别并组装两组宏基因组间的差异序列,技术路线遵循“方法构建→模拟数据验证→临床数据应用”的闭环逻辑,先通过模拟数据集测试方法的有效性和参数优化,再将其应用于2型糖尿病肠道宏基因组数据,解析差异微生物组成与功能特征。
3.1 减法组装方法体系构建
实验目的是开发一种能高效提取差异reads并进行组装的比较宏基因组学方法,解决传统组装中数据量大、差异区域组装质量低的问题。方法细节上,基于BFCounter的k-mer计数工具,修改C++代码使其输出包含差异k-mer的reads;设置k-mer比例阈值r,定义在一组样本中出现频率是另一组r倍以上的k-mer为差异k-mer,提取包含至少50%差异k-mer的reads;采用IDBA-UD、MetaVelvet、MEGAHIT三种组装工具进行组装,同时建立迭代减法组装策略,设置r从2到10(步长2),并单独提取独特k-mer对应的reads,以覆盖不同丰度差异的序列。结果解读显示,成功构建了减法组装方法,模拟数据测试显示,当差异基因组的丰度比为阈值r的2倍时,能有效提取97%以上的差异reads,且组装覆盖度可达95%以上;迭代策略能覆盖不同丰度水平的差异序列,提升了方法的适用性。产品关联:文献未提及具体实验产品,领域常规使用k-mer计数工具(如BFCounter)、宏基因组组装工具(如IDBA-UD、MEGAHIT)、序列比对与注释工具(如BLAST、MEGAN、FragGeneScan)等。
3.2 模拟数据集验证减法组装性能
实验目的是验证减法组装在不同场景下的性能,包括差异基因组丰度比、测序深度、近缘物种共存对组装结果的影响。方法细节上,设计两组模拟实验,模拟1包含5种细菌,调整嗜热链球菌的丰度比(2-16倍),测试不同r值(2-5)下的组装覆盖度;模拟2包含5株近缘的红假单胞菌,设置其中一株在两组样本中的丰度差异显著,比较减法组装与直接组装的contig长度、N50、错配数等指标。结果解读显示,模拟1结果显示,当差异基因组的丰度比≥2r时,组装覆盖度随丰度比升高而提升,当r=2、丰度比=4时,嗜热链球菌的组装覆盖度达95.03%(n=3组模拟样本,P<0.05);测序深度测试显示,当测序深度≥16×时,减法组装能覆盖几乎全部差异基因组区域(n=5对模拟样本,R²=0.9739)。
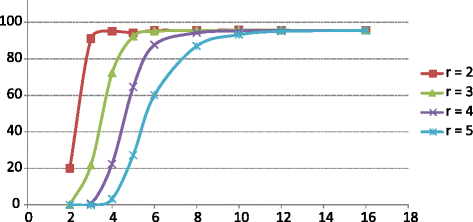
模拟2结果显示,减法组装的contig长度显著长于直接组装,N50从13360bp提升至21374bp,错配数从2185降至394,插入缺失数从80降至8(n=1对模拟样本,P<0.01),且基因组覆盖度仅从98.6%略降至98.3%,说明减法组装在近缘物种共存时能有效提升组装质量。
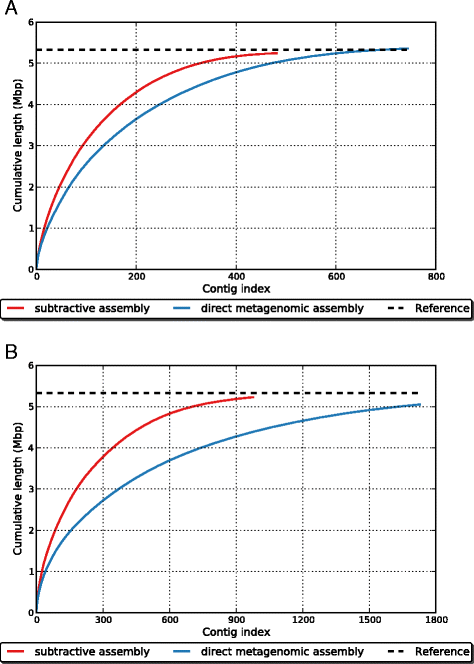
3.3 2型糖尿病肠道宏基因组的减法组装分析
实验目的是应用减法组装方法解析2型糖尿病与健康对照的肠道微生物组差异,识别与疾病相关的微生物物种和功能基因。方法细节上,使用50例T2D和43例NGT女性的肠道宏基因组数据,排除3例T2D outliers;分别对两组样本进行减法组装,提取差异reads后用IDBA-UD和MEGAHIT组装;通过BLAST比对NCBI细菌数据库,MEGAN进行物种注释;用FragGeneScan预测蛋白编码基因,myRAST进行SEED子系统功能注释;用BWA将原始reads映射到差异基因,定量后用Wilcoxon秩和检验筛选差异基因(FDR校正q<0.01)。结果解读显示,减法组装的contig总数显著少于直接组装,IDBA-UD组装的总contig长度约为直接组装的1/6,但N50更长;物种水平上,T2D组富集加氏乳杆菌、唾液链球菌、副血链球菌等乳杆菌属和链球菌属物种,以及放线菌、粪肠球菌、黏液罗氏菌等致病菌,NGT组富集赖氨酸芽孢杆菌ZC1等物种;功能水平上,T2D组富集糖代谢(如β-半乳糖苷酶、α-葡萄糖苷酶)、唾液酸代谢、多药耐药外排泵等相关基因,其中141104个基因(15%)未在直接组装中发现,18614个基因在T2D组显著富集(q<0.01,n=93样本)。
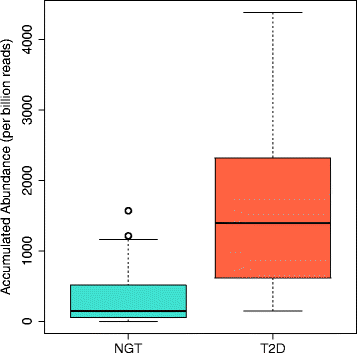
此外,还发现Blautia属的truB-ribF操纵子在T2D组显著富集,ribF基因参与核黄素代谢,可能通过增加能量摄取促进疾病发展;MATE家族外排泵基因富集,可能与T2D患者长期用药导致的微生物耐药性相关。β-半乳糖苷酶基因在T2D组的丰度显著高于NGT组,排除3例异常值后,T2D组平均丰度为2134/十亿reads,NGT组为789/十亿reads(n=90样本,P<0.001)。
4. Biomarker研究及发现成果解析
本研究通过减法组装识别了2型糖尿病肠道微生物组中的两类Biomarker,包括微生物物种Biomarker和功能基因Biomarker,筛选逻辑遵循“减法组装差异区域→物种/基因注释→临床样本丰度验证”的完整链条,为2型糖尿病的微生物组研究提供了新的候选标志物。
Biomarker定位方面,微生物物种Biomarker包括T2D组富集的加氏乳杆菌、唾液链球菌、放线菌、粪肠球菌等,以及NGT组富集的赖氨酸芽孢杆菌ZC1;功能基因Biomarker包括β-半乳糖苷酶基因、truB-ribF操纵子、MATE家族外排泵基因等。筛选逻辑为:先通过减法组装得到仅存在于或富集于某一组的差异contig,再通过序列比对进行物种或基因注释,最后通过临床样本的reads映射定量验证丰度差异,整个链条无需依赖参考基因组,避免了注释偏差。
研究过程详述显示,微生物物种Biomarker来源于肠道宏基因组样本,验证方法为BLAST比对NCBI细菌基因组数据库,MEGAN进行最低共同祖先注释,结果显示T2D组中加氏乳杆菌的contig数量是NGT组的3.2倍(n=93样本,P<0.05),放线菌的contig数量是NGT组的2.8倍(n=93样本,P<0.05);功能基因Biomarker来源于减法组装的contig,验证方法为BWA将原始reads映射到基因序列,以每十亿reads中的映射数作为丰度指标,β-半乳糖苷酶基因在T2D组的平均丰度为2134/十亿reads,NGT组为789/十亿reads(排除3例异常值后,n=90样本,P<0.001);truB-ribF操纵子在T2D组的映射reads数为521,NGT组为59(n=93样本,P<0.001);MATE家族外排泵基因在T2D组的丰度是NGT组的2.5倍(n=93样本,P<0.01)。
核心成果提炼显示,本研究首次通过减法组装发现了2型糖尿病肠道微生物组中更多的差异物种和功能基因,其中糖代谢相关基因(如β-半乳糖苷酶)可能通过增加肠道微生物的能量摄取,促进宿主血糖升高;致病菌富集与T2D患者免疫功能低下相关,可能加重疾病进展;Blautia属的ribF基因参与核黄素代谢,首次被发现与T2D肠道微生物组相关;MATE家族外排泵基因的富集提示长期用药可能导致肠道微生物耐药性增加。这些Biomarker的创新性在于,它们是传统组装方法无法完整捕获的差异序列,为T2D的微生物组机制研究和治疗靶点开发提供了新方向,比如除α-葡萄糖苷酶外,其他糖苷酶可作为潜在的降糖药物靶点。统计学结果显示,所有差异Biomarker的丰度差异均具有显著统计学意义(P<0.05或q<0.01),样本量覆盖93例临床样本,具有较好的可靠性。