1. 领域背景与文献引入
文献英文标题:Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs;发表期刊:Biomarker Research;影响因子:未公开;研究领域:肿瘤精准医学与DNA修复缺陷生物标志物研究
领域共识:肿瘤精准医学的核心是基于肿瘤分子特征匹配个性化治疗方案,其中DNA修复缺陷是重要的治疗靶点方向。2005年BRCA1/2缺陷与PARP抑制剂的合成致死效应被发现,开启了DNA修复缺陷靶向治疗的时代;后续研究证实BRCA1/2缺陷的卵巢癌、三阴性乳腺癌对铂类化疗和PARP抑制剂高度敏感,且这类肿瘤会积累大量基因组瘢痕(如拷贝数变异、杂合性缺失等)。当前领域热点集中于开发泛癌种的DNA修复缺陷生物标志物,以识别更多潜在的敏感人群,但核心问题在于:现有基因组瘢痕标志物仅在少数癌种中得到验证,缺乏泛癌层面的系统分析,无法明确非铂类标准治疗癌种中是否存在DNA修复缺陷亚群。
针对这一研究空白,本研究通过泛癌队列分析三种已发表的基因组瘢痕标志物(端粒等位基因失衡NtAI、大规模染色体转变LST、同源重组缺陷杂合性缺失HRD-LOH)的分布特征,旨在识别潜在的铂类或PARP抑制剂敏感人群,为现有抗癌药物拓展新适应症提供依据,具有重要的临床转化价值。
2. 文献综述解析
作者对领域内现有研究的分类维度主要为“DNA修复缺陷类型”与“生物标志物开发方向”,结构化总结如下:在DNA修复缺陷与治疗敏感性研究方面,已有明确结论显示BRCA1/2缺陷的卵巢癌、三阴性乳腺癌对铂类化疗和PARP抑制剂敏感,其技术优势在于基于基因缺陷的精准分层,但局限性是仅覆盖少数癌种,且约20%的敏感肿瘤无BRCA1/2突变;在基因组瘢痕标志物研究方面,NtAI、LST、HRD-LOH三种标志物分别在三阴性乳腺癌、卵巢癌中被证实可预测铂类敏感性,技术优势是基于SNP阵列数据可量化分析,无需基因测序,但局限性是缺乏泛癌数据支持,且三种标志物的相关性与生物学重叠性未被系统研究。
通过对比现有研究的未解决问题,本研究的创新价值凸显:首次在泛癌层面系统分析三种基因组瘢痕标志物的分布特征,明确其在非铂类标准治疗癌种中的高得分亚群,弥补了现有研究仅聚焦少数癌种的不足,为泛癌种DNA修复缺陷人群的识别提供了新的分析框架。
3. 研究思路总结与详细解析
本研究的核心目标是明确三种同源重组缺陷相关基因组瘢痕标志物在泛癌中的分布特征,核心科学问题是“泛癌中是否存在未被识别的DNA修复缺陷亚群,可从铂类或PARP抑制剂治疗中获益”,技术路线遵循“数据获取→标志物量化→分布与关联分析→结论”的闭环逻辑。
3.1 泛癌队列数据收集与预处理
实验目的是获取标准化的泛癌基因组与临床数据,为后续标志物分析提供高质量数据集。方法上,研究纳入了癌症基因组图谱(TCGA)中15种癌种的5371例肿瘤样本,收集Affymetrix SNP6基因分型芯片数据与对应的临床注释信息;采用Aroma Affymetrix CRMAv2算法进行芯片数据预处理,通过CalMaTe和TumorBoost算法校正等位基因特异性拷贝数偏差,使用ASCAT算法分析肿瘤倍体与纯度,排除肿瘤细胞纯度低于36%的样本。结果显示,最终得到5371例符合质量控制标准的样本数据集,可用于后续的标志物量化与分析。文献未提及具体实验产品,领域常规使用Affymetrix SNP6基因分型芯片、R语言生物信息学分析工具包(如Aroma.Affymetrix、ASCAT等)。
3.2 三种基因组瘢痕标志物的量化
实验目的是统一量化NtAI、LST、HRD-LOH三种基因组瘢痕标志物,确保分析方法的一致性与准确性。方法上,基于R语言实现三种标志物的量化算法,同时对原方法进行修正:针对NtAI算法,引入倍体校正逻辑,避免非整倍体样本的得分偏差;针对HRD-LOH算法,未排除17号染色体,以保留泛癌分析中的潜在信息;LST算法则严格遵循原文献的实现标准。结果显示,成功得到所有样本的三个标志物得分,且得分分布符合预期的生物学特征(如卵巢癌样本得分显著高于其他癌种)。文献未提及具体实验产品,领域常规使用R语言基础包与自定义生物信息学分析脚本。
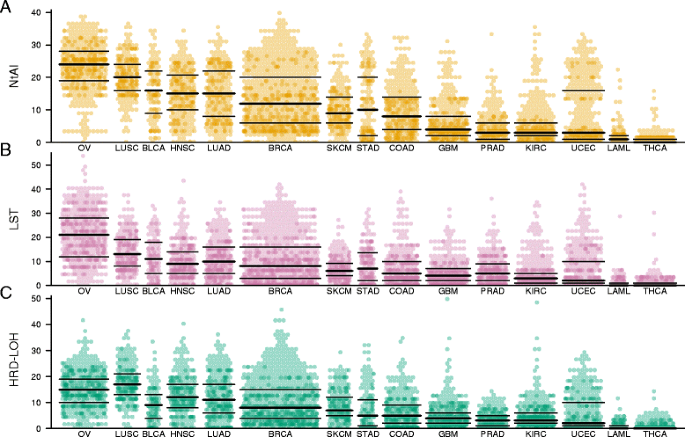
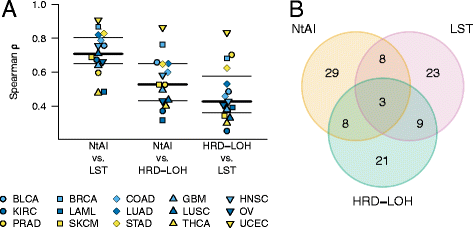
3.3 标志物泛癌分布与相关性分析
实验目的是分析三种标志物在不同癌种的分布特征及相互关联,明确其生物学重叠性。方法上,采用箱线图展示三种标志物在15种癌种的得分分布,通过Spearman秩相关分析评估三个标志物得分的相关性,使用韦恩图分析三种标志物所计数的基因组瘢痕事件的重叠率。结果显示,卵巢癌与肺鳞癌的三个标志物得分中位数最高(卵巢癌NtAI=24、LST=20、HRD-LOH=15),急性髓系白血病与甲状腺癌得分最低;三个标志物之间具有良好的相关性,Spearman相关系数ρ为0.73-0.87,但单个基因组瘢痕事件的重叠率较低(仅3.1%的事件被三个标志物同时计数)。这表明三种标志物虽测量不同类型的基因组瘢痕,但反映的是相似的DNA修复缺陷状态。文献未提及具体实验产品,领域常规使用R语言可视化工具包(如ggplot2、VennDiagram等)。
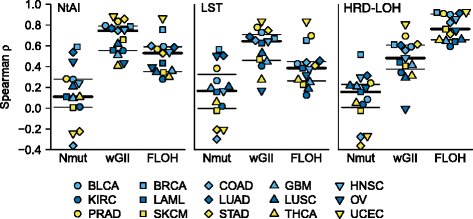
3.4 标志物与基因组不稳定及临床特征的关联分析
实验目的是探索三种标志物与其他基因组不稳定指标、临床特征的关联,明确其生物学意义与临床价值。方法上,对比三种标志物与加权基因组完整性指数(wGII)、杂合性缺失频率(FLOH)、总突变数(Nmut)的相关性;分析肿瘤倍体、p53突变状态、分期分级、吸烟史与标志物得分的关联。结果显示,NtAI与LST与wGII相关性较高(中位数ρ=0.75、0.64),HRD-LOH与FLOH相关性最高(中位数ρ=0.76);近四倍体样本的NtAI与LST得分显著高于近二倍体样本(11/15、12/15癌种P<0.05);p53突变样本的三个标志物得分显著高于野生型样本(多数癌种P<0.05,Fisher精确检验OR=3.64,P<0.0001);部分癌种中高分期/分级、吸烟人群的标志物得分更高。文献未提及具体实验产品,领域常规使用R语言统计分析工具包(如stats、survival等)。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究涉及的Biomarker为三种同源重组缺陷相关基因组瘢痕标志物:端粒等位基因失衡(NtAI)、大规模染色体转变(LST)、同源重组缺陷杂合性缺失(HRD-LOH),其筛选与验证逻辑为“已发表标志物算法优化→泛癌队列量化分析→与临床特征及治疗相关性关联验证”,形成了完整的生物标志物研究链条。
研究过程详述
三种Biomarker的来源为TCGA泛癌队列的SNP6基因分型芯片数据,验证方法为生物信息学量化分析与关联分析:通过修正后的算法量化所有样本的三个标志物得分,分析其在不同癌种的分布,以及与p53突变、倍体等临床特征的关联。特异性与敏感性方面,在已验证的卵巢癌与三阴性乳腺癌中,三个标志物可有效识别铂类敏感人群(文献引用既往研究数据);在泛癌分析中,不同癌种的标志物得分差异显著,如子宫内膜癌中25%的样本呈现高得分(NtAI 16-33,LST 10-45,HRD-LOH 10-29),提示这部分样本可能存在DNA修复缺陷。
核心成果提炼
本研究的核心成果包括:1. 首次泛癌证实三种基因组瘢痕标志物在多数癌种中存在高得分亚群,包括非铂类标准治疗的癌种(如子宫内膜癌、肾癌等),这些亚群可能对铂类化疗或PARP抑制剂敏感;2. 明确三种标志物具有良好的相关性,反映相似的DNA修复缺陷状态,但测量的基因组瘢痕事件重叠率低,提示可联合使用以提高识别准确性;3. 发现p53突变、近四倍体状态与三种标志物得分显著相关,为DNA修复缺陷的机制研究提供了线索。统计学结果方面,三个标志物的Spearman相关系数为0.73-0.87,p53突变样本与野生型样本的标志物得分差异具有统计学意义(多数癌种P<0.05,Fisher精确检验OR=3.64,P<0.0001)。推测:这些高得分亚群患者可作为铂类或PARP抑制剂临床研究的潜在入组人群,需进一步临床验证其治疗敏感性。