1. 领域背景与文献引入
文献英文标题:Antileukemic potential of methylated indolequinone MAC681 through immunogenic necroptosis and PARP1 degradation;发表期刊:Biomarker Research;影响因子:未公开;研究领域:慢性髓性白血病靶向治疗、免疫原性细胞死亡、PARP1分子调控
慢性髓性白血病(CML)是由t(9;22)染色体易位产生的BCR-ABL融合基因驱动的造血系统恶性肿瘤,2001年第一代酪氨酸激酶抑制剂(TKI)伊马替尼的获批开启了CML靶向治疗的里程碑,二代、三代TKIs的应用进一步提升了患者的无进展生存期,但约20%的患者会出现耐药或治疗不耐受,白血病干细胞(LSCs)依赖线粒体氧化磷酸化(OXPHOS)的代谢重编程及DNA修复缺陷是耐药的关键机制。当前研究热点聚焦于靶向LSCs的代谢脆弱性、诱导非凋亡细胞死亡及激活抗肿瘤免疫,但仍缺乏同时靶向DNA修复缺陷、代谢重编程及免疫原性的多维度治疗策略,且PARP1在CML中的靶向价值尚未被充分挖掘。
针对上述研究空白,本研究聚焦CML细胞的BRCA样DNA修复缺陷与代谢重编程特征,系统探究甲基化吲哚醌MAC681的抗白血病作用及机制,通过诱导PARP1降解、代谢灾难和免疫原性坏死,为耐药CML的治疗提供新的靶点与联合治疗范式。
2. 文献综述解析
作者围绕“CML治疗瓶颈-机制靶点-药物潜力”三层逻辑构建综述框架,先梳理CML的治疗现状与耐药机制,再聚焦LSCs的代谢特征与DNA修复缺陷,最后总结吲哚醌类化合物的抗癌潜力及免疫原性细胞死亡(ICD)的临床价值,系统整合现有研究的优势与局限。
现有研究显示,一线TKIs可有效抑制BCR-ABL激酶活性,但无法根除依赖OXPHOS的LSCs,且耐药细胞会通过代谢重编程维持存活;靶向代谢的策略如2-脱氧葡萄糖可增强TKIs疗效,但单一靶向代谢易出现代偿性耐药;PARP抑制剂在BRCA缺陷肿瘤中已获批应用,但CML细胞的BRCA样DNA修复缺陷尚未被充分利用于靶向治疗;吲哚醌类化合物具有广谱抗癌活性,但针对CML的特异性作用及ICD诱导能力的研究较少,缺乏对“PARP1降解-免疫原性坏死”关联机制的探索。
本研究突破现有单一靶向的局限,首次发现MAC681可同时靶向PARP1降解、代谢灾难及ICD诱导,填补了CML中该关联机制的研究空白,且验证了与第三代TKI asciminib的协同作用,为耐药CML的联合治疗提供了新的实验依据与理论支撑。
3. 研究思路总结与详细解析
本研究以“CML细胞的代谢与DNA修复缺陷→MAC681的细胞毒性及机制→PARP1降解的分子机制→免疫原性验证→联合治疗潜力”为闭环研究逻辑,通过生物信息学分析、细胞实验、动物实验多维度验证MAC681的抗白血病作用,实验设计覆盖“靶点筛选-机制验证-体内外功能-临床转化潜力”的完整学术链条。
3.1 CML患者转录组特征分析
实验目的:明确CML干细胞的代谢重编程与DNA修复缺陷的分子特征。方法细节:整合公共GEO数据集(GSE5550、GSE47927_GSE97562、GSE13159),采用R语言limma包进行差异表达分析,通过Reactome、GO、GSEA进行通路富集,对比CML患者与健康供体CD34+细胞的基因表达差异。结果解读:转录组分析显示CML干细胞显著富集细胞周期、代谢、DNA修复通路,其中BRCA突变肿瘤相关基因特征富集,提示CML细胞存在双链DNA修复缺陷;PARP1在CML干细胞中表达显著高于健康对照(GSE5550及GSE47927_GSE97562数据集,p≤0.0001),且高PARP1表达与线粒体功能通路正相关;NAD+代谢通路基因在CML干细胞中显著富集(Fisher精确检验p<0.05)。
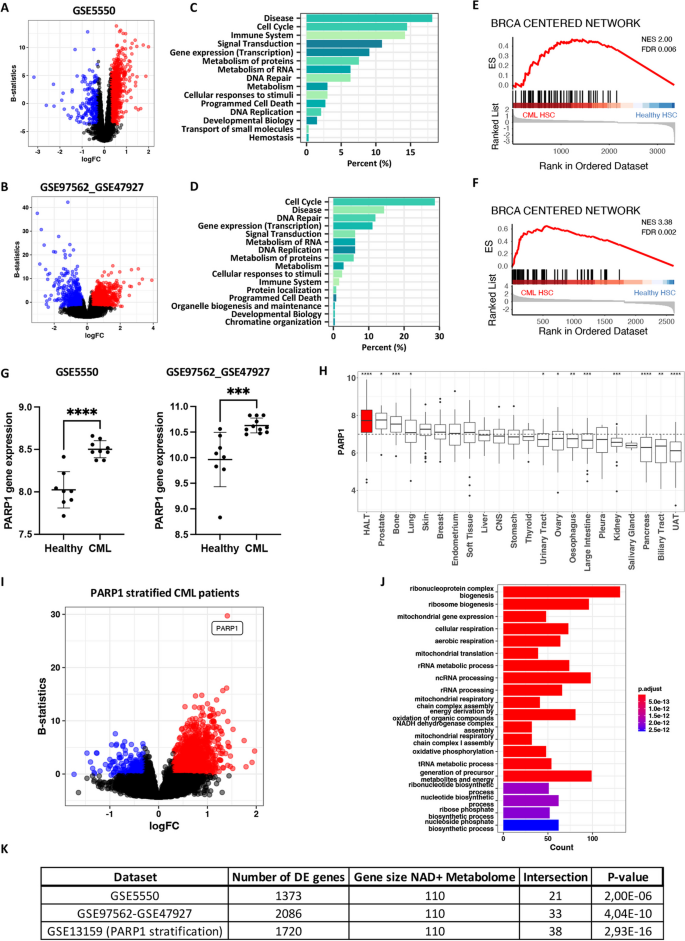
产品关联:文献未提及具体实验产品,领域常规使用R语言生物信息学分析工具(如limma、fgsea、ggplot2包)、公共基因表达数据库(GEO、CCLE)。
3.2 MAC681诱导CML细胞DNA损伤与代谢灾难
实验目的:验证MAC681对CML细胞的细胞毒性及代谢干扰机制。方法细节:采用γH2AX免疫荧光及流式检测DNA双链损伤,Seahorse分析仪检测ATP生成速率,NAD/NADH-Glo试剂盒检测NAD+与NADH水平,透射电镜(TEM)观察线粒体形态,流式细胞术检测线粒体膜电位、钙稳态。结果解读:MAC681处理K-562细胞1h后,γH2AX阳性细胞占比达38.3%,2-8h升至76-86%(n=3,p<0.0001);NAD+水平2h内下降87%,ATP生成速率2h内降低43%(n=3,p<0.01);TEM显示线粒体肿胀、嵴断裂,流式检测到线粒体膜电位时间依赖性丧失及钙稳态紊乱,提示代谢灾难的发生。
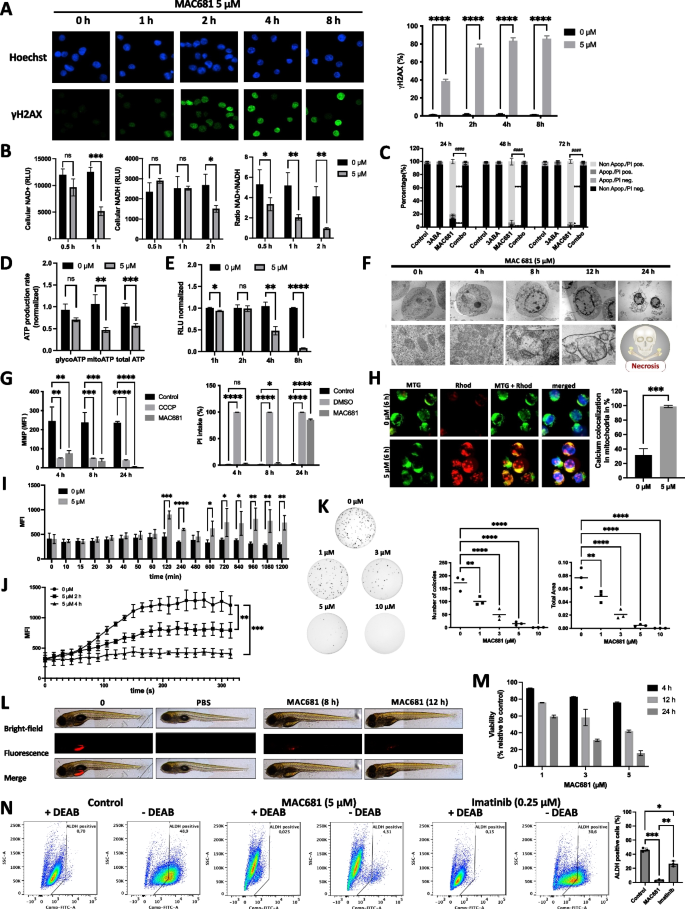
产品关联:实验所用关键产品包括Seahorse ATP Real-Time rate assay kit(Agilent)、NAD/NADH-Glo™ Assay(Promega)、MitoTracker Red(Molecular Probes/Invitrogen)、透射电镜(Philips CM12)。
3.3 PARP1降解机制与3-氨基苯甲酰胺的保护作用
实验目的:探究MAC681诱导PARP1降解的分子机制及3-氨基苯甲酰胺(3ABA)的干预效应。方法细节:Western blot检测PARP1蛋白水平,分子对接(PatchDock、AutoDock4)模拟MAC681与PARP1的结合,质谱检测3ABA与MAC681的加合物形成,流式细胞术检测细胞死亡与DNA损伤。结果解读:MAC681处理8h后,K-562细胞中全长PARP1(116kDa)完全消失,且无凋亡切割片段;3ABA预处理可阻止PARP1降解及DNA损伤(n=3,p<0.0001);分子对接显示MAC681结合PARP1催化区(结合能-7.1kcal/mol),3ABA与MAC681形成的加合物结合更紧密(结合能-9.4、-8.5kcal/mol),质谱证实6位加合物的存在,提示3ABA通过化学结合抑制MAC681的DNA损伤活性。
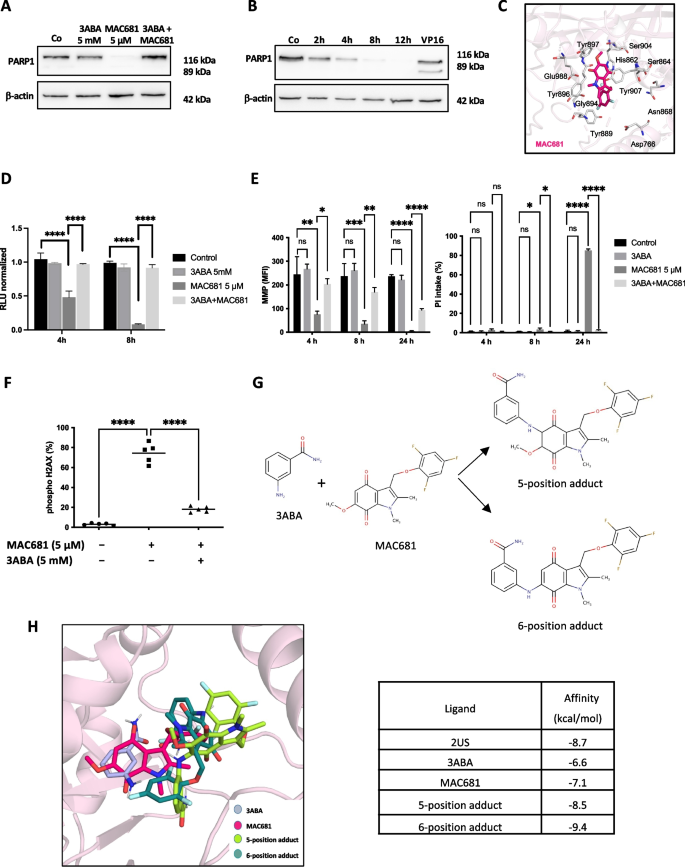
产品关联:实验所用关键产品包括Western blot相关试剂(ECL Plus Western Blotting Detection System Kit,GE Healthcare)、分子对接软件(AutoDock4、PatchDock)、Agilent 6410 triple quadrupole质谱仪。
3.4 MAC681诱导免疫原性细胞死亡的验证
实验目的:确认MAC681诱导的细胞死亡具有免疫原性,可激活抗肿瘤免疫。方法细节:检测损伤相关分子模式(DAMPs):ATP分泌(ENLITEN® ATP Assay)、HMGB1释放(ELISA)、钙网蛋白(CRT)表面表达(流式);巨噬细胞吞噬实验(J774A1与K-562共培养);C57BL/6小鼠疫苗实验(MAC681处理的C1498细胞接种后挑战活细胞)。结果解读:MAC681处理后,K-562细胞ATP分泌增加2-4倍(n=3,p<0.001),CRT表面表达翻倍(n=3,p<0.05),HMGB1释放4h后显著升高(n=3,p<0.05);巨噬细胞对MAC681处理细胞的吞噬率提升160倍(n=3,p<0.01);小鼠疫苗接种后,肿瘤体积较对照组显著降低(n=每组多只,p<0.0001),证实ICD的诱导能力。
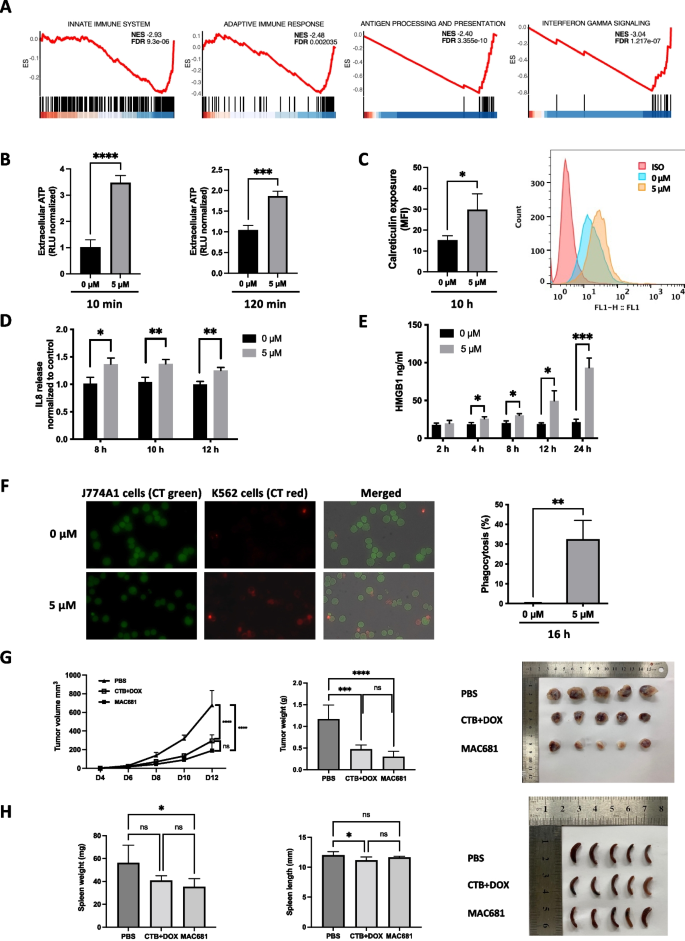
产品关联:实验所用关键产品包括HMGB1 ELISA kit(IBL International)、ENLITEN® ATP Assay System(Promega)、calreticulin抗体(Abcam, 2907)、CellTracker染料(Thermofisher Scientific)。
3.5 MAC681与asciminib的协同作用研究
实验目的:验证MAC681与第三代TKI asciminib联合对敏感及耐药CML细胞的治疗潜力。方法细节:采用台盼蓝排斥实验检测细胞活力,Chou-Talalay算法计算联合指数(CI),评估K-562(敏感)及K-562R(耐药)细胞的协同效应。结果解读:MAC681与asciminib联合处理后,K-562R细胞在24h、48h、72h的联合指数均<1,K-562细胞在多组浓度组合下CI<1,证实显著协同作用。
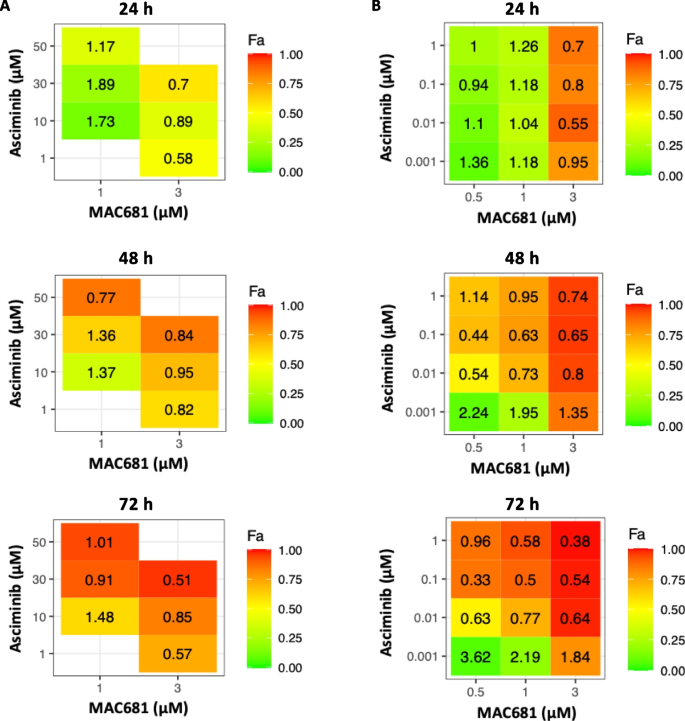
产品关联:文献未提及具体实验产品,领域常规使用台盼蓝染色试剂、Compusyn协同分析软件。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究将PARP1定位为CML的关键治疗靶点及潜在预后Biomarker,筛选逻辑为:通过公共转录组数据集分析发现CML干细胞中PARP1高表达,且与BRCA样DNA修复缺陷、线粒体代谢通路正相关;验证逻辑为细胞系实验验证MAC681通过降解PARP1发挥抗白血病作用,临床样本关联其表达与CML的分子特征,形成“生物信息学筛选-细胞功能验证-临床样本关联”的完整逻辑链。
研究过程详述
PARP1的来源为CML患者CD34+干细胞及CML细胞系(K-562、K-562R等);验证方法包括qRT-PCR、Western blot检测表达水平,分子对接验证结合能力,细胞实验验证功能;特异性数据显示CML患者PARP1表达显著高于健康对照(GSE5550及GSE47927_GSE97562数据集,p≤0.0001),敏感性方面,高PARP1表达的CML细胞对MAC681的细胞毒性更敏感(n=3,p<0.05)。
核心成果提炼
PARP1作为CML的功能Biomarker,其高表达与CML细胞的代谢重编程、DNA修复缺陷直接相关,文献未明确提及风险比(HR)数据;MAC681通过降解PARP1诱导代谢灾难与免疫原性坏死,为耐药CML提供新的治疗靶点;创新性在于首次在CML中证实“PARP1降解-免疫原性坏死”的关联机制,且验证了与asciminib的协同治疗潜力,为CML的免疫代谢联合治疗提供了新的实验依据,有望推动耐药CML治疗策略的优化。