1. 领域背景与文献引入
文献英文标题:Evaluating topography of mutational signatures with SigProfilerTopography;发表期刊:Genome Biology;影响因子:未公开;研究领域:肿瘤基因组学、突变特征与基因组拓扑学交叉领域
体细胞突变是癌症和正常体细胞基因组的普遍特征,由内源性和外源性诱变过程塑造,每个过程具有特征性的突变模式即突变特征。领域共识:突变在基因组中的分布并非随机,多数突变特征受人类基因组拓扑特征影响,包括复制时序、染色质状态、组蛋白修饰、转录因子结合位点等,还可能表现出转录链、复制链不对称性及链协同诱变。现有研究已开发多种突变特征分析工具,但缺乏能全面整合基因组拓扑特征与突变特征活性的计算工具,无法系统解析诱变过程与基因组结构的相互作用,这一空白限制了对突变特征生物学机制的深入理解。本文针对这一问题,开发了SigProfilerTopography工具,旨在全面评估染色质组织、组蛋白修饰、转录因子结合、DNA复制与转录等对不同突变过程活性的影响,揭示突变特征的独特拓扑特性及其潜在生物学机制。
2. 文献综述解析
作者按工具功能与拓扑分析能力对现有研究进行分类,将突变特征分析工具分为仅关注突变模式本身、部分整合拓扑特征两类。现有研究中,多数工具如SigProfilerExtractor等专注于突变特征的提取与归因,未涉及拓扑特征分析;少数工具如MutationalPatterns、TensorSignatures、Mutalisk虽考虑部分拓扑特征,但存在明显局限性:Mutalisk仅分析样本中所有体细胞突变的拓扑分布,未区分不同突变特征的独特拓扑行为;MutationalPatterns可比较基因组不同区域的突变模式,但未考虑基因组结构、突变特征的模式及活性,统计分析缺乏针对性;TensorSignatures在提取突变特征时整合部分拓扑特征,但性能逊于传统工具,且拓扑分析能力有限。此外,现有工具普遍不支持用户自定义多种实验来源的拓扑数据,也未覆盖所有小突变类型(如双碱基替换、小插入缺失)。通过对比现有工具的未解决问题,本研究的创新点在于首次开发了全面整合基因组拓扑特征与突变特征活性的自动化工具,支持用户自定义ATAC-seq、Repli-seq等多种实验来源的拓扑数据格式,覆盖单碱基替换、双碱基替换、小插入缺失所有小突变类型,并通过模拟突变构建统计检验的零假设,系统解析突变特征的拓扑偏好,为揭示诱变过程的生物学机制提供了全新的技术范式。
3. 研究思路总结与详细解析
整体框架为开发SigProfilerTopography开源Python包,以突变数据和拓扑特征数据为输入,通过模拟突变、突变特征归因、统计对比真实与模拟突变的拓扑分布,揭示不同突变特征的基因组拓扑偏好,核心科学问题是解析诱变过程与基因组结构的相互作用机制,技术路线遵循“工具开发→多维度拓扑分析→实验验证→机制阐释”的闭环逻辑。
3.1 工具开发与计算流程设计
实验目的是构建能全面分析突变特征拓扑分布的计算工具,明确其核心功能与计算逻辑。方法细节:工具以Python开发,支持VCF、MAF等多种突变数据格式,以及wig、bigWig、bed等多种拓扑特征数据格式;默认利用SigProfilerAssignment将突变归因于COSMIC数据库的已知突变特征,也支持用户自定义突变特征的模式与活性;通过SigProfilerSimulator模拟100次真实突变,保持突变类型与染色体分布特征;仅保留归因概率≥90%的突变进行后续统计分析,采用z检验、Fisher精确检验等方法对比真实与模拟突变的拓扑分布,经Benjamini-Hochberg校正多重检验。结果解读:工具实现了从数据输入到多维度拓扑分析的全流程自动化,输出包括占据分析、链不对称性、复制时序、链协同诱变等多种结果,能有效区分真实突变与模拟突变的拓扑分布差异,揭示突变特征的独特拓扑行为。产品关联:实验所用关键工具:SigProfilerSimulator、SigProfilerAssignment、SigProfilerMatrixGenerator(均为AlexandrovLab开发的开源Python包)。
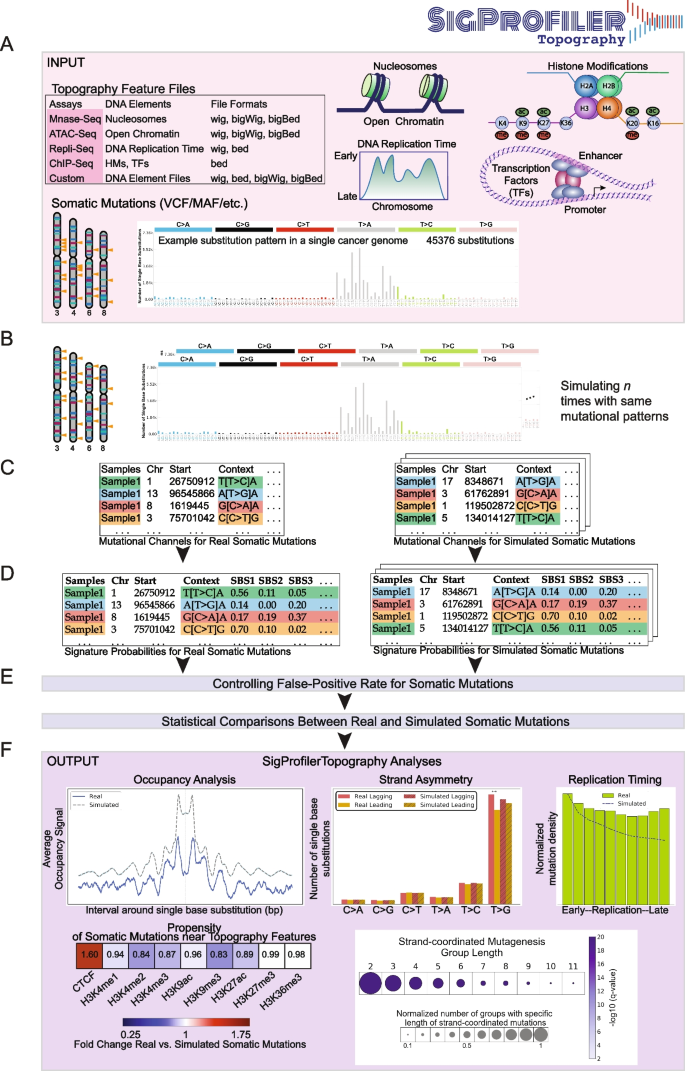
3.2 特征占据分析
实验目的是评估突变特征在拓扑特征附近的分布模式,明确其与染色质结构、转录因子结合的关联。方法细节:以±1kb突变侧翼区域为分析窗口,整合微球菌核酸酶测序(MNase-seq)、染色质免疫沉淀测序(ChIP-seq)等拓扑数据,计算真实与模拟突变在侧翼区域的平均信号强度,通过统计检验对比两者的富集/缺失模式;同时支持全基因组与局部区域两种零假设模型,优化统计检验的准确性。结果解读:对552例食管鳞癌(n=552)队列的分析显示,单碱基替换特征SBS17b突变在核小体区域表现出约190碱基对的周期性分布,与核小体位置的损伤修复差异相关,且在CTCF结合位点显著富集(q值≤0.05);插入缺失特征ID2突变在核小体占据区域缺失,在CTCF结合位点富集(q值≤0.05);采用局部背景模型后,单碱基替换特征SBS1在CTCF和H3K4me1区域的缺失效应更显著,从全基因组背景的0.82倍和0.93倍降至0.35倍和0.70倍(q值≤0.05),SBS17b在CTCF区域的富集从1.74倍提升至2.79倍(q值≤0.05),凸显了局部基因组背景对突变分布的影响。产品关联:文献未提及具体实验试剂,领域常规使用MNase-seq试剂盒、ChIP-seq试剂盒进行拓扑特征检测。
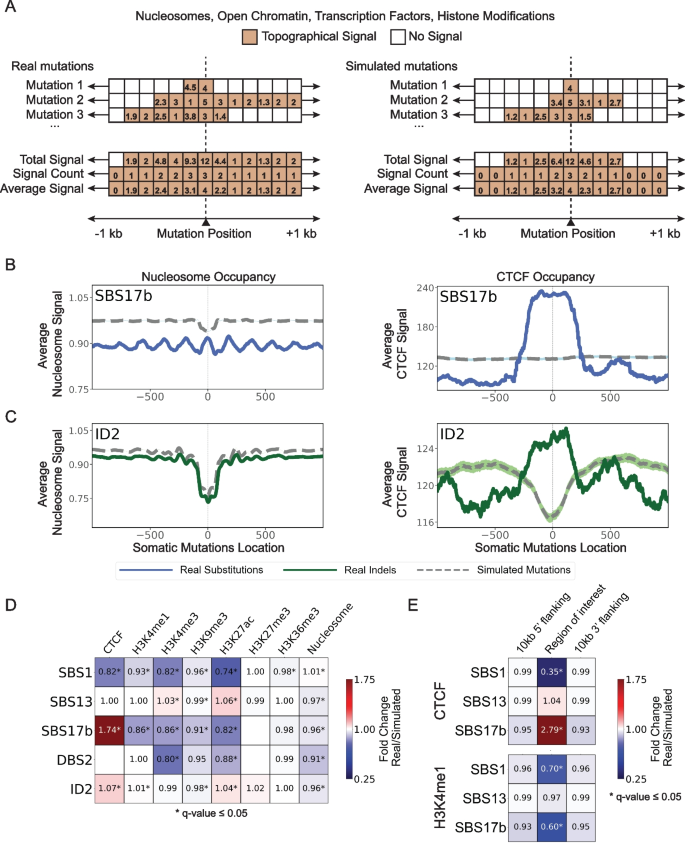
3.3 复制时序与复制链不对称性分析
实验目的是解析突变特征与DNA复制过程的关联,明确其在复制时序区域及复制链上的分布偏好。方法细节:基于复制测序(Repli-seq)数据定义早/晚复制区域,将复制时序信号分为10个分位数,计算突变特征在每个分位数的归一化突变密度;通过小波变换识别复制起始/终止区域,定义前导链与滞后链,统计突变特征在两条链上的分布差异,采用Fisher精确检验进行显著性分析。结果解读:单碱基替换特征SBS2和SBS17b突变的归一化突变密度从早复制到晚复制区域逐渐增加(q值≤0.05),与晚复制区域损伤修复效率较低的领域共识一致;SBS2突变在滞后链显著富集(q值≤0.05),符合APOBEC脱氨酶靶向复制过程中滞后链单链DNA的特性,而SBS17b未表现出显著的复制链不对称性。产品关联:文献未提及具体实验试剂,领域常规使用Repli-seq试剂盒进行复制时序检测。
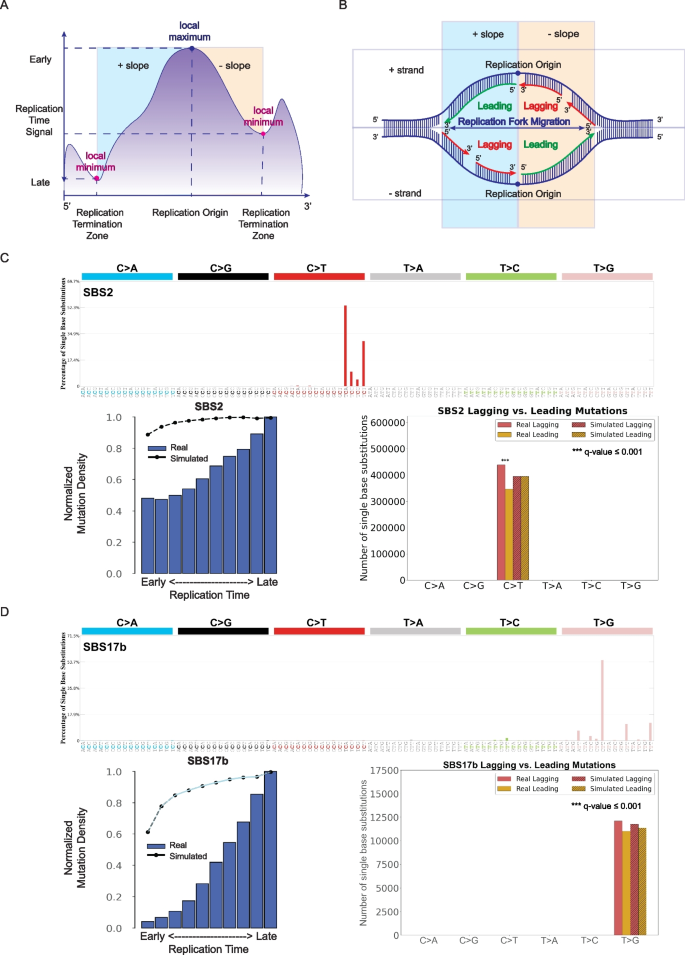
3.4 转录链不对称性分析
实验目的是评估突变特征与转录过程的关联,明确其在转录链、非转录链及基因区/基因间区的分布偏好。方法细节:将基因区突变分为转录链与非转录链,基因间区突变单独分类,统计突变特征在不同区域的分布,对比真实与模拟突变的差异,采用Fisher精确检验进行显著性分析。结果解读:单碱基替换特征SBS16突变在转录链的T>C突变显著富集,且在基因区富集(q值≤0.05,n=552),与酒精诱导的转录偶联损伤机制一致;SBS17b未表现出显著的转录链不对称性,但在基因间区显著富集(q值≤0.05,n=552),提示其诱变过程可能优先作用于非编码区域。产品关联:文献未提及具体实验试剂,领域常规使用RNA-seq数据辅助定义转录区域。
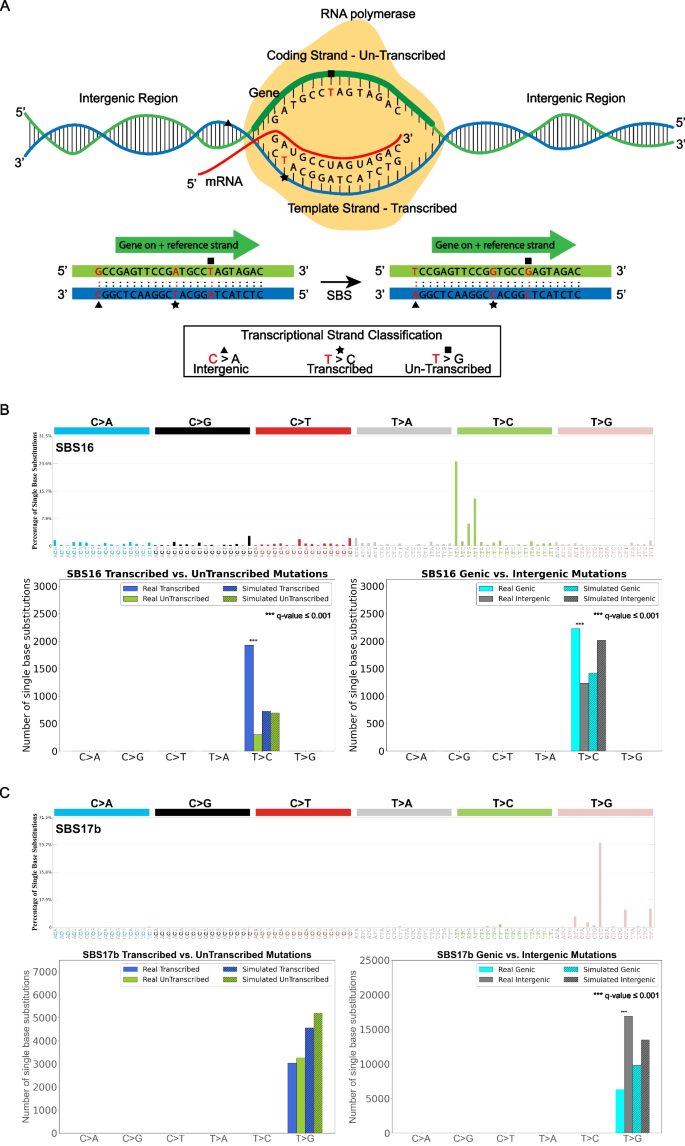
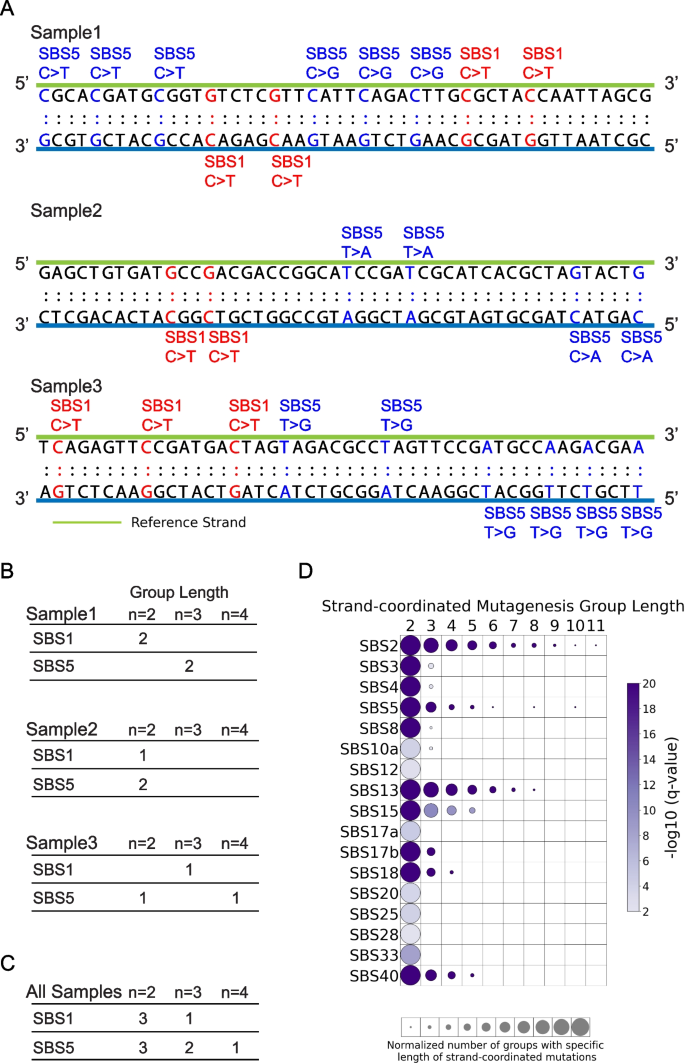
3.5 链协同诱变分析
实验目的是解析突变特征的局部簇状分布模式,明确链协同诱变的发生机制。方法细节:将同一链上距离≤10kb、归因于同一突变特征的连续单碱基替换定义为链协同诱变组,统计真实与模拟突变中不同长度组的数量,采用z检验进行显著性分析。结果解读:对食管鳞癌队列的分析显示,APOBEC相关特征SBS2和SBS13存在长达11个连续突变的链协同诱变组(q值≤0.05,n=552),符合APOBEC诱导的kataegis现象;SBS5和SBS40等“平坦”特征也表现出不同长度的链协同诱变;多数突变特征存在长度为2的突变簇,可能与omikli事件相关。产品关联:文献未提及具体实验试剂,领域常规使用全基因组测序数据进行突变簇检测。
4. Biomarker研究及发现成果
Biomarker定位为突变特征的基因组拓扑偏好特征,筛选验证逻辑为通过真实突变与模拟突变的统计对比,排除随机分布的影响,验证拓扑偏好的显著性,采用全基因组与局部背景两种模型优化检验准确性。研究过程:以552例食管鳞癌全基因组测序数据为研究对象(n=552),结合ATAC-seq、Repli-seq、MNase-seq、ChIP-seq等多组学拓扑数据,分析8种拓扑特征与5种突变特征的相互作用,通过z检验、Fisher精确检验等方法进行显著性分析,经Benjamini-Hochberg校正多重检验。核心成果:揭示了多个突变特征的独特拓扑行为,包括单碱基替换特征SBS17b在CTCF结合位点富集(局部背景下富集倍数2.79,q值≤0.05)、在H3K4me1区域缺失(局部背景下缺失倍数0.60,q值≤0.05);SBS2在晚复制区域富集、在滞后链富集(q值≤0.05);SBS16在转录链富集、在基因区富集(q值≤0.05);APOBEC相关特征存在显著的链协同诱变(q值≤0.05)。这些成果为解析突变特征的生物学机制提供了新的视角,例如SBS17b的核小体周期性分布提示其诱变过程与核小体位置的DNA损伤修复差异相关,SBS2的滞后链富集支持APOBEC脱氨酶靶向复制过程中产生的单链DNA的机制。