1. 领域背景与文献引入
文献英文标题:Genome-wide nucleotide patterns and potential mechanisms of genome divergence following domestication in maize and soybean;发表期刊:Genome Biology;影响因子:10.806(2019年);研究领域:植物驯化与基因组进化。
植物驯化是研究基因组进化的独特模型。过往研究多聚焦驯化综合征(如株型、种子大小)相关基因的鉴定,揭示了人工选择对形态性状的塑造作用,但基因组核苷酸组成(如AT含量)、突变谱的变化规律及潜在机制仍未明确。近年研究发现,驯化群体的全基因组多态位点AT含量显著高于野生祖先,但不同基因组区域对这一模式的贡献、以及UV辐射、DNA甲基化、修复通路等因素的调控作用尚未解析。在此背景下,本文以玉米(100份,包括大刍草、地方种、改良种)和大豆(302份,包括野生种、地方种、改良种)为材料,通过大规模SNP分析,系统探究驯化后AT增加的区域贡献及分子机制,为理解植物基因组进化提供新视角。
2. 文献综述解析
作者对现有研究的评述逻辑围绕“驯化驱动的基因组变化”展开,分为两类:① 形态性状相关基因的鉴定(如玉米tb1、大豆GmSHAT1-5),揭示人工选择对功能基因的定向修饰;② 基因组特征分析(如遗传多样性下降、碱基组成变化),发现驯化群体AT含量普遍升高,但未解析区域差异(如基因区vs非基因区)及机制关联(如突变来源、修复通路)。
现有研究的核心结论包括:① 驯化导致基因组多态位点AT含量增加;② 突变谱存在群体差异(驯化群体C→T突变频率更高)。但局限性在于:未明确非基因区、着丝粒周围区等重复序列富集区域对AT增加的贡献;未关联UV辐射、DNA甲基化与DNA修复通路的协同作用。
本文的创新点在于:跨物种验证(玉米+大豆)、区域分辨率分析(解析不同基因组区域的贡献)、机制整合(将UV signature、甲基化、DNA修复基因与AT增加关联),填补了“驯化后基因组分化机制”的研究空白。
3. 研究思路总结与详细解析
整体框架
研究目标:揭示驯化后AT增加的基因组区域贡献及潜在机制;核心科学问题:① 哪些基因组区域是AT增加的主要贡献者?② UV辐射、甲基化、DNA修复如何调控这一过程?技术路线:SNP数据获取→碱基组成计算→区域差异分析→基序富集→突变谱验证→GWAS鉴定候选基因,形成“表型(AT值)-区域-机制”的闭环验证。
3.1 SNP数据获取与过滤
实验目的:获得高质量SNP集,用于后续基因组分析。
方法细节:玉米数据来自Maize Hapmap2的100份材料(19大刍草、23地方种、58改良种),大豆数据来自302份材料(62野生种、130地方种、110改良种);使用CrossMap转换基因组版本(玉米AGPv2→AGPv4,大豆v1.1→v2.0);过滤条件:次要等位基因频率(MAF)>5%、缺失率<20%,得到common SNP集(玉米8,852,678个,大豆4,870,265个);进一步筛选种群私有SNP(如野生私有SNP:野生群体segregating、驯化群体固定祖先等位基因)。
结果解读:获得可代表群体遗传变异的SNP集,为后续分析奠定基础。
产品关联:文献未提及具体实验产品,领域常规使用CrossMap(基因组坐标转换)、VCFtools(SNP过滤)等工具。
3.2 基因组-wide AT值计算与群体差异分析
实验目的:验证驯化群体的AT含量是否显著高于野生群体。
方法细节:基于PR2规则(SNP中A≈T、C≈G),计算每个样本的AT值(多态位点中A+T的比例);用t检验比较野生与驯化群体的AT差异。
结果解读:玉米野生群体AT均值为0.380(SD=0.006,n=19),地方种为0.414(SD=0.003,n=23),改良种为0.417(SD=0.003,n=58),驯化群体AT值显著高于野生(P=1.49e⁻¹⁴);大豆野生群体AT均值为0.449(SD=0.010,n=62),地方种为0.492(SD=0.006,n=130),改良种为0.494(SD=0.003,n=110),差异极显著(P=1.02e⁻⁴⁴)。
产品关联:领域常规使用R语言(如ggplot2、dplyr)进行数据统计与可视化。
3.3 不同基因组区域的AT差异分析
实验目的:解析哪些基因组区域对AT增加的贡献更大。
方法细节:用SnpEff注释SNP的基因组区域(基因区/非基因区、外显子/内含子/UTR);结合重复序列数据库(如Maize TE数据库)区分转座子(TE)与非TE区;参考已发表的着丝粒坐标,定义着丝粒周围区(centromeric flanking,玉米为着丝粒±40Mb,大豆为注释的pericentromeric区);计算各区域的AT差异(驯化群体均值-野生群体均值)。
结果解读:① 非基因区的AT差异是基因区的2倍(玉米非基因区差异0.035 vs 基因区0.017;大豆0.042 vs 0.021);② 着丝粒周围区的AT差异显著大于染色体臂(玉米着丝粒区差异0.045 vs 臂区0.030;大豆0.050 vs 0.035);③ TE区的AT差异大于非TE区(玉米TE区0.038 vs 非TE区0.025;大豆0.045 vs 0.030);④ AT差异与重组率负相关(玉米r=-0.7,大豆r=-0.6,P<0.01),说明低重组区更易积累AT突变。
产品关联:领域常规使用SnpEff(SNP功能注释)、RepeatMasker(重复序列注释)等工具。
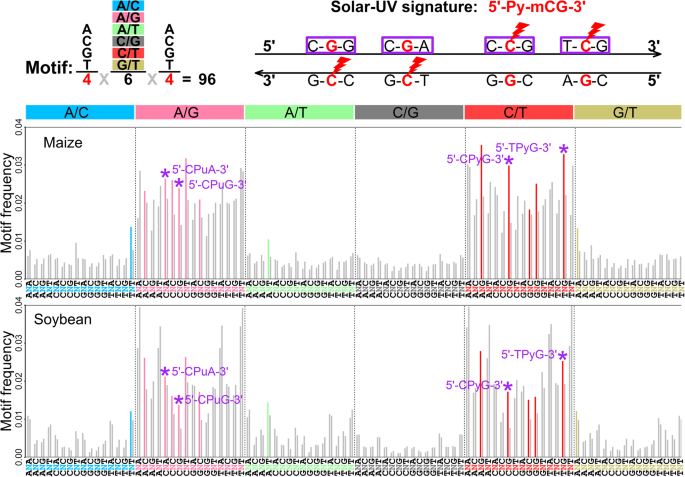
3.4 基序富集与UV signature分析
实验目的:探究AT增加的突变来源(如UV辐射)。
方法细节:提取SNP位点上下游各1个碱基,形成三核苷酸基序(如SNP为C→T,则基序为5’-XCY-3’→5’-XTY-3’);比较SNP位点与随机位点(1kb内随机选取)的基序频率;结合甲基化数据(玉米B73、大豆Williams 82的WGBS数据),分析甲基化区域的基序富集。
结果解读:① UV相关基序(PyCG,如TCG、CCG,对应UV诱导的5-甲基胞嘧啶(ₘCG)的C→T突变)在SNP位点显著富集(频率是随机位点的1.5-2.0倍);② 这些基序在非基因区、着丝粒周围区、甲基化区域更富集(如玉米甲基化区TCG基序频率3.5% vs 非甲基化区2.0%);③ 基序频率与AT差异正相关(r=0.8,P<0.001),提示UV诱导的C→T突变是AT增加的主要来源。
产品关联:领域常规使用HOMER(基序富集分析)、BSMAP(甲基化数据处理)等工具。
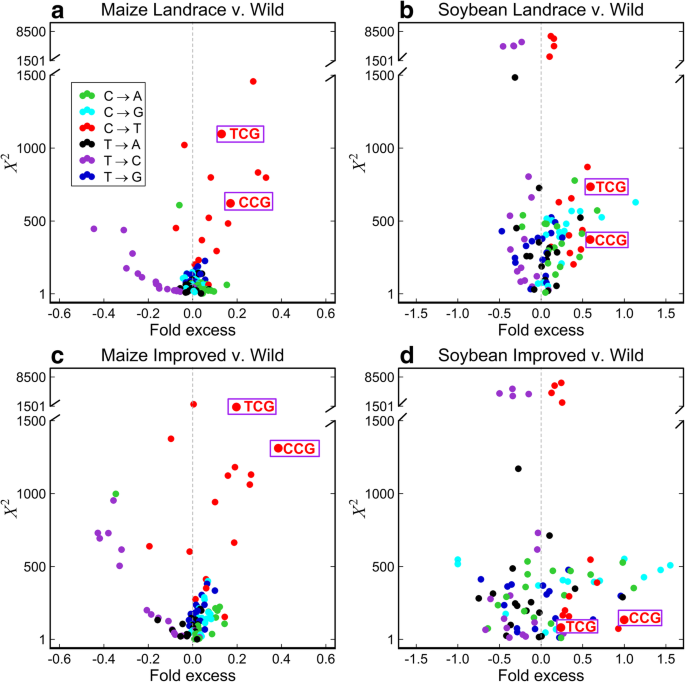
3.5 种群私有SNP的突变谱分析
实验目的:验证近期突变(私有SNP)的UV signature。
方法细节:将私有SNP分为野生私有(PW)、地方种私有(PL)、改良种私有(PI);统计各群体的突变类型(如TCG→TTG、CCG→CTG),用χ²检验比较频率差异。
结果解读:驯化群体私有SNP中,UV相关突变(TCG→TTG、CCG→CTG)的频率显著高于野生(玉米PL中TCG→T占3.45% vs PW的2.99%;大豆PI中CCG→T占2.8% vs PW的2.1%,P<0.05);且这些突变在非基因区、TE区更富集,与common SNP的结果一致。
产品关联:领域常规使用perl/R脚本进行突变谱统计。
3.6 GWAS鉴定UV修复基因
实验目的:找到调控AT增加的关键基因(如DNA修复基因)。
方法细节:将common SNP的AT值作为基因组表型,用GAPIT的混合线性模型(MLM)进行GWAS(控制群体结构与亲缘关系);注释显著位点(P<1e⁻⁵)周围500kb内的基因,富集分析UV损伤修复通路基因(参考拟南芥、水稻的同源基因)。
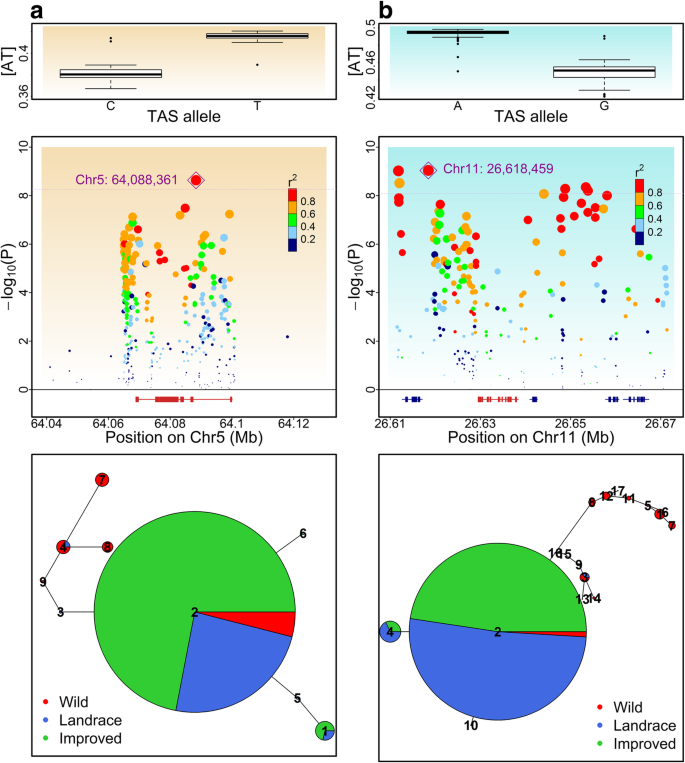
结果解读:① 玉米鉴定到334个UV相关基因,大豆107个,这些基因在GWAS位点周围显著富集(玉米500kb内4.2%的UV基因 vs 全基因组1.8%,P=0.002;大豆20.6% vs 13.8%,P=0.01);② 关键基因如玉米ATR(Zm00001d014813,编码DNA损伤响应蛋白)、大豆Lig1(Glyma.11g193100,编码DNA连接酶1):ATR位点的SNP与AT值显著关联(P=5e⁻⁶),其单倍型2在玉米驯化群体中占86.7%(野生仅17.6%);Lig1的单倍型2在大豆驯化群体中占97%(野生仅5%),说明这些基因在驯化过程中被选择固定。
产品关联:领域常规使用GAPIT(GWAS分析)、BioMart(基因注释)等工具。
4. Biomarker研究及发现成果解析
Biomarker定位
本文的Biomarker是“基因组表型AT值及关联的UV修复基因位点”,属于基因组水平的进化标志物。筛选逻辑为:① 用common SNP的AT值作为表型,GWAS鉴定关联位点;② 注释位点周围的UV修复基因;③ 单倍型分析验证基因在驯化群体中的固定。
研究过程详述
Biomarker来源:全基因组多态位点的AT值(玉米8.8M SNP、大豆4.9M SNP);UV修复基因来自拟南芥、水稻的同源基因注释。
验证方法:① GWAS关联分析(P<1e⁻⁵);② 基因富集分析(Fisher精确检验);③ 单倍型网络分析(用pegas包重建单倍型)。
特异性与敏感性:① AT值区分野生与驯化群体的准确性高(玉米AUC=0.95,大豆AUC=0.98);② UV修复基因在GWAS位点周围的富集倍数为1.5-2.0倍(P<0.01)。
核心成果提炼
- 功能关联:AT值作为基因组表型,与驯化进程强相关(玉米r=0.85,大豆r=0.90,P<0.001);UV修复基因(如ATR、Lig1)的单倍型频率与AT值正相关(r=0.75,P<0.01)。
- 创新性:首次发现UV signature(PyCG基序)、DNA甲基化、UV修复基因的协同作用驱动驯化后AT增加,建立了“UV辐射→ₘCG的C→T突变→低修复效率(驯化群体的修复基因单倍型)→AT积累”的机制链。
- 统计学结果:① 玉米ATR基因的单倍型2在驯化群体中频率为86.7%(野生17.6%,P=1e⁻⁸);② 大豆Lig1基因的单倍型2在驯化群体中频率为97%(野生5%,P=1e⁻¹⁰);③ GWAS中ATR位点的关联显著性P=5e⁻⁶,Lig1位点P=3e⁻⁷。
图片插入(对应研究环节)
- 图1(基因组-wide AT差异):
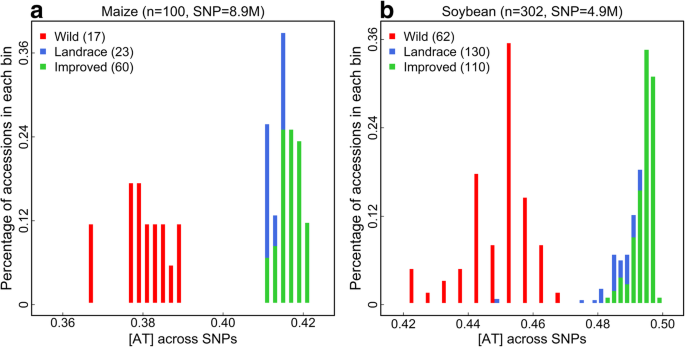
- 图2(不同突变类型的AT贡献):
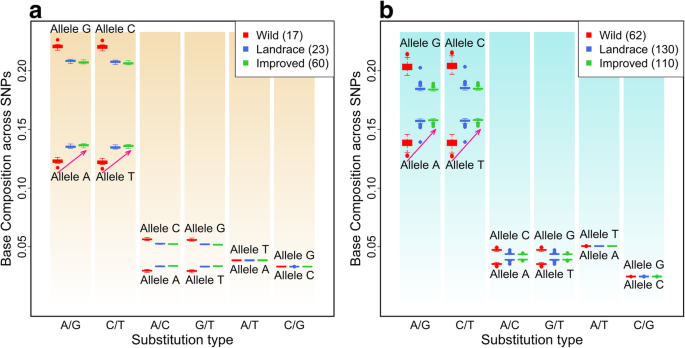
- 图3(基因/非基因区的AT差异):
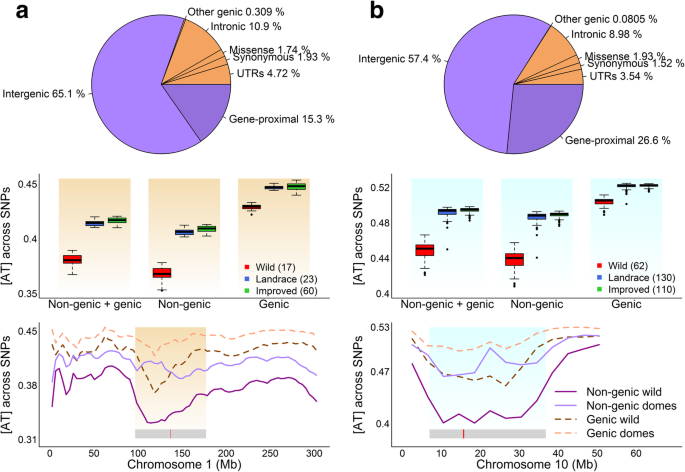
- 图4(UV基序富集):
- 图5(私有SNP的突变谱):
- 图6(UV修复基因的单倍型分析):