1. 领域背景与文献引入
文献英文标题:An integrative probabilistic model for identification of structural variation in sequencing data;发表期刊:Genome Biology;影响因子:2012年具体数值未公开,该期刊为基因组学与生物信息学领域顶级期刊;研究领域:基因组结构变异检测(生物信息学与基因组学交叉领域)。
结构变异(Structural Variation, SV)包括DNA片段的缺失、重复、倒位、易位等类型,是人类正常基因组遗传差异及肿瘤基因组体细胞变异的重要组成部分。2000年后,随着DNA微阵列和下一代测序技术的发展,学界对结构变异的认知迅速提升,下一代测序技术已广泛应用于正常基因组遗传变异及肿瘤基因组体细胞结构变异的检测。然而,下一代测序的短读长和短插入片段特性,使得结构变异的检测面临诸多挑战,尤其是重复序列区域的结构变异,由于读段比对的不确定性,现有检测方法的灵敏度和特异性难以兼顾。领域共识:现有结构变异检测方法通常仅利用测序数据中的单一信号,如读深度(Read Depth, RD)、配对读(Paired Read, PR)或拆分读(Split Read, SR),且多数方法忽略存在多比对结果的读段,导致对重复区域结构变异的检测灵敏度显著降低,同时假阳性率较高。
针对上述领域痛点,本研究旨在开发一种整合多种测序信号并能处理多比对读段的结构变异检测方法,提出了GASVPro算法,该算法将配对读信号和读深度信号(包括断点区域的局部读深度信号)整合到统一的概率模型中,通过马尔可夫链蒙特卡洛(Markov Chain Monte Carlo, MCMC)方法处理存在多比对结果的读段,最终实现结构变异检测灵敏度与特异性的同步提升,为重复区域结构变异的检测提供了新的技术方案。
2. 文献综述解析
作者对现有结构变异检测研究按所利用的测序信号类型进行分类评述,系统梳理了不同方法的优势与局限性,并明确了本研究的创新定位。
现有结构变异检测方法可分为三类,第一类是基于读深度信号的方法,这类方法通过比对读段在基因组区域的覆盖深度变化检测拷贝数变异,优势在于能有效识别大片段拷贝数变异,局限性是无法检测倒位、易位等平衡结构变异;第二类是基于配对读信号的方法,通过比对读段的预期与实际映射位置差异检测结构变异,优势在于能覆盖所有类型的结构变异,局限性是依赖读段的唯一比对结果,在重复序列区域的检测灵敏度较低,且多数方法仅以支持读段数量作为判断依据,假阳性率较高;第三类是基于拆分读信号的方法,通过直接识别跨越结构变异断点的读段检测变异,优势在于能精准定位断点,局限性是受下一代测序短读长的限制,应用范围较窄。此外,现有研究中仅有少数方法在后期处理阶段整合两种信号,无法从根本上提升检测灵敏度,且均未有效处理存在多比对结果的读段,导致重复区域的结构变异检测性能不佳。
本研究的核心创新点在于首次将配对读信号与读深度信号(包括断点局部读深度信号)整合到统一的概率模型中,而非仅在后期处理阶段进行信号整合,同时通过MCMC方法处理存在多比对结果的读段,解决了重复区域结构变异检测的痛点。与现有方法相比,GASVPro在删除检测的特异性上提升50%至90%,在倒位检测的特异性上提升50%,同时保持较高的检测灵敏度,填补了多信号整合与多比对读段处理的结构变异检测方法空白。
3. 研究思路总结与详细解析
本研究的核心目标是开发一种高灵敏度、高特异性的结构变异检测方法,核心科学问题是如何通过整合多种测序信号并处理多比对读段,提升重复区域结构变异的检测性能,技术路线遵循“模型构建→算法实现→模拟数据验证→真实数据验证→性能比较”的闭环逻辑,最终验证了GASVPro算法的优越性。
3.1 结构变异检测概率模型构建
实验目的:构建整合配对读与读深度信号的概率模型,实现结构变异的精准检测,并量化断点位置的不确定性。
方法细节:基于已发表的GASV算法的断点多边形表示方法,定义断点读深度(breakend Read Depth, beRD)信号,针对结构变异的三种状态(纯合结构变异、杂合结构变异、非结构变异)建立概率模型,分别计算不同状态下的似然概率;针对删除型结构变异,优化模型以利用整个删除区域的读深度信号提升检测性能。
结果解读:通过似然比判断结构变异的真实性,模型能够有效区分真实结构变异与假阳性结果,同时量化断点位置的不确定性;图1展示了配对读信号、断点多边形表示、读深度与断点读深度信号的差异,直观呈现了不同类型结构变异的测序信号特征。
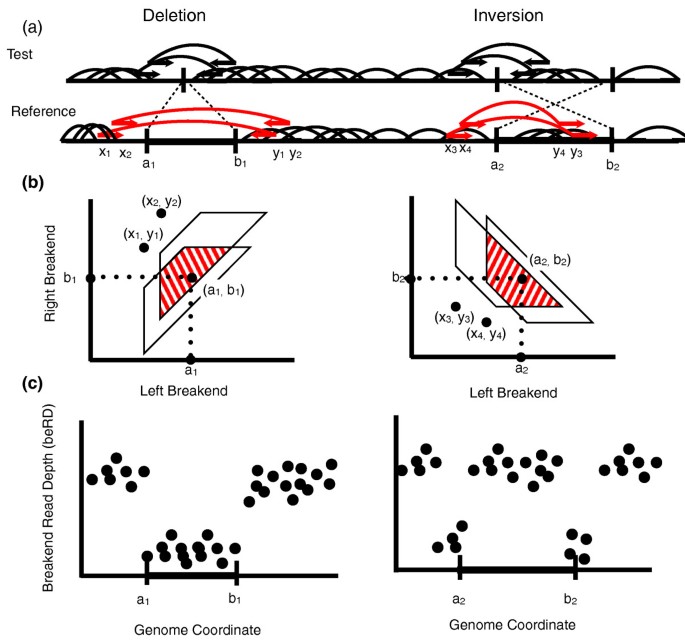
产品关联:文献未提及具体实验产品,领域常规使用BWA、Novoalign等序列比对软件,SAMtools等测序数据分析工具。
3.2 多比对读段的MCMC处理策略
实验目的:解决重复序列区域读段多比对结果导致的结构变异检测假阳性问题,提升重复区域的检测灵敏度。
方法细节:构建比对矩阵与映射矩阵,采用Metropolis Hastings算法设计马尔可夫链,定义四种移动类型(Naive移动、移除列移动、恢复列移动、交换列移动)实现映射矩阵的采样,将全局采样分解为独立连通分量的并行采样,提升计算效率。
结果解读:MCMC方法能够从读段的多比对结果中选择真实的映射位置,有效降低重复区域结构变异检测的假阳性率,同时提升检测灵敏度;图2展示了GASVPro算法的整体流程,清晰呈现了多比对读段的处理过程,
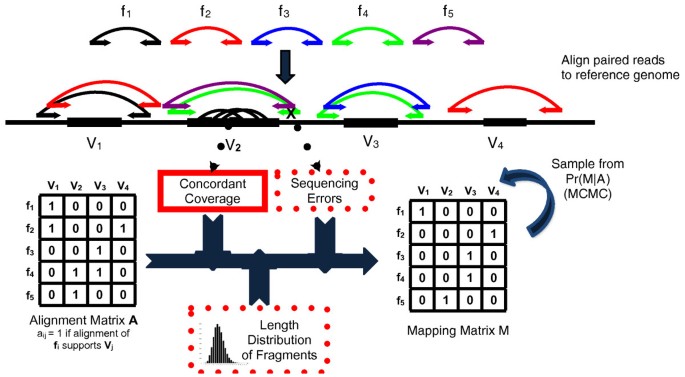
图3展示了连通分量的并行采样策略,说明算法的高效性。
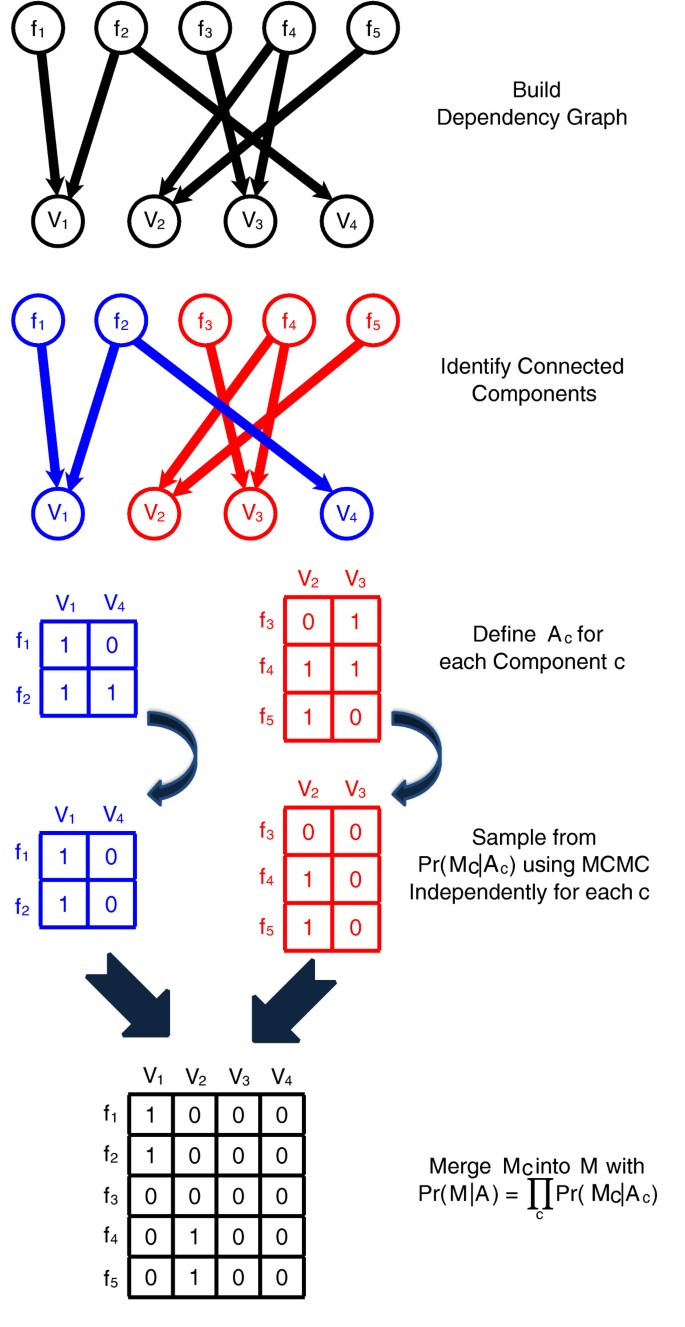
产品关联:文献未提及具体实验产品,领域常规使用Python、C++等编程语言实现算法,采用MCMC相关工具包进行采样。
3.3 模拟数据与真实基因组数据验证
实验目的:验证GASVPro算法的检测性能,并与现有主流结构变异检测方法进行比较。
方法细节:构建模拟数据集,将Venter基因组17号染色体的已知结构变异插入人类参考基因组,模拟100×覆盖度的配对读测序数据;采用NA12878和NA18507两个人类基因组的真实测序数据,统一比对与后处理流程,采用“double uncertainty”指标比较GASVPro与BreakDancer、Hydra、GASV、CNVer等方法的检测性能。
结果解读:模拟数据验证显示,GASVPro在删除检测的特异性上比现有方法提升50%至90%,能够检测所有4个已知倒位结构变异;真实数据验证显示,在NA12878的Pilot2数据中,GASVPro检测到119个已知删除结构变异,仅产生907个新预测,假阳性率比GASV降低95%;图4展示了模拟数据中删除检测的ROC曲线,直观呈现GASVPro的性能优势,
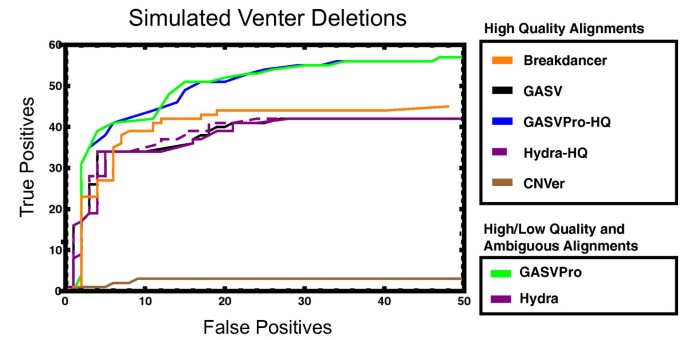
图5展示了NA12878数据中删除检测的结果比较,
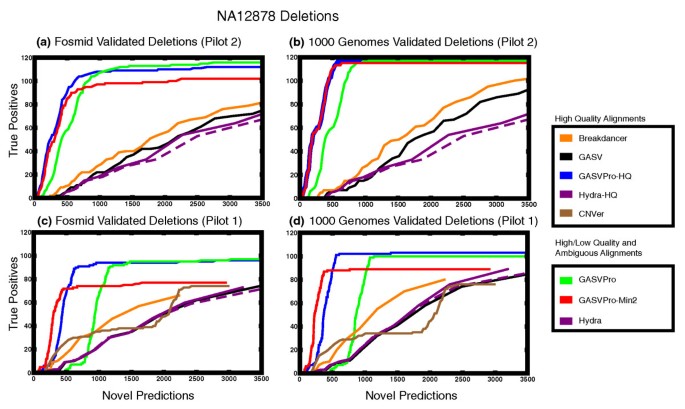
图6展示了NA18507数据中删除检测的结果比较,
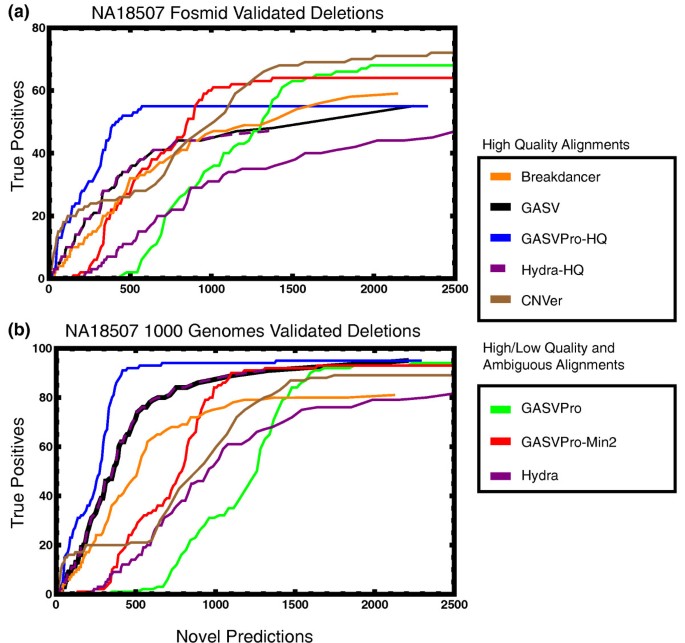
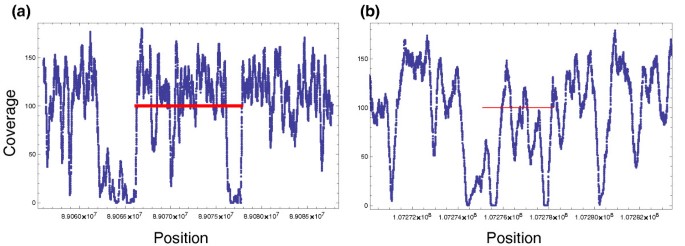
图7展示了NA18507数据中倒位结构变异的断点读深度信号,验证了该信号的有效性。
产品关联:文献未提及具体实验产品,领域常规使用1000 Genomes Project公开测序数据,采用已发表的结构变异验证集进行比较。
4. Biomarker研究及发现成果
本研究检测的生物标志物为基因组中的删除、倒位等结构变异类型,通过模拟数据与真实数据的验证,证明GASVPro算法能够高效、精准地检测这类生物标志物,为基因组结构变异的研究提供了新的技术手段。
Biomarker定位:本研究检测的生物标志物为基因组中的删除、倒位等结构变异类型,筛选与验证逻辑为:在模拟数据中插入已知结构变异,验证算法的检测能力;在真实数据中与已通过fosmid测序、1000 Genomes Project验证的结构变异集进行比较,验证算法的实际性能。
研究过程详述:结构变异的来源包括模拟构建的变异基因组与真实人类基因组的测序数据,验证方法采用“double uncertainty”指标,将算法预测的结构变异与已知验证集进行比对,量化检测的特异性与敏感性。特异性方面,GASVPro在删除结构变异检测中的假阳性结果比现有方法减少50%至90%,在倒位结构变异检测中减少50%;敏感性方面,模拟数据中能够检测所有4个已知倒位结构变异,真实数据中在NA12878个体的Pilot2数据中检测到119个已知删除结构变异(共139个),在NA18507个体中检测到55个已知删除结构变异(共93个)。
核心成果提炼:GASVPro算法通过整合多种测序信号与处理多比对读段,显著提升了结构变异检测的灵敏度与特异性,尤其是在重复序列区域的结构变异检测性能上表现突出,为基因组结构变异的检测提供了新的技术范式。研究未提供具体的P值统计结果,但ROC曲线与性能比较数据显示,GASVPro的检测性能显著优于现有主流方法,为后续结构变异的功能研究与临床应用奠定了方法学基础。