1. 领域背景与文献引入
文献英文标题:Characterization and modeling of the Haemophilus influenzae core and supragenomes based on the complete genomic sequences of Rd and 12 clinical nontypeable strains;发表期刊:Genome Biology;影响因子:2007年约10.23;研究领域:细菌比较基因组学、感染性疾病(非分型流感嗜血杆菌)
流感嗜血杆菌是定植于人类鼻咽部的革兰氏阴性菌,分为有荚膜(a-f血清型)和无荚膜的非分型流感嗜血杆菌(NTHi),其中b型有荚膜株(Hib)曾是儿童脑膜炎的主要致病菌,1980年代Hib疫苗推广后,发达国家的Hib感染已近乎消除,而NTHi成为急性中耳炎、慢性阻塞性肺疾病(COPD)、鼻窦炎等呼吸道感染的主要致病菌。领域发展关键节点包括1995年Rd株(首个完成全基因组测序的自由生活细菌)的测序,2005年Tettelin等提出细菌泛基因组(supragenome)概念并构建无乳链球菌泛基因组模型。当前研究热点聚焦于细菌泛基因组的结构与功能、水平基因转移对致病性的调控、慢性感染的基因组基础。未解决的核心问题包括NTHi泛基因组的大小与结构尚不明确,核心基因组与非核心基因组的功能差异及来源未系统解析,现有泛基因组模型的均等概率采样假设是否适用于具有自然转化能力的细菌(如NTHi的DNA摄取依赖特异性信号序列)。
结合领域现状,此前针对NTHi的比较基因组研究样本量较小(通常仅3-5株),无法准确反映物种层面的基因组多样性,因此本研究通过测序12株临床NTHi株并结合已发表的Rd株,共13株样本开展大规模比较基因组分析,构建有限泛基因组模型以验证分布式基因组假说(DGH),为NTHi的致病性研究、诊断标志物开发提供理论基础。
2. 文献综述解析
作者以基因的分布特征(核心、分布式、独特)与来源(外源同源性、密码子使用模式)为分类维度,系统梳理了细菌泛基因组研究的现状与NTHi基因组多样性的前期研究,明确了现有研究的局限性并提出本研究的创新方向。
现有泛基因组研究中,Tettelin等2005年针对无乳链球菌的研究构建了泛基因组模型,认为其泛基因组为无限大小,但该模型假设非核心的contingency genes被均等概率采样,未考虑细菌自然转化的特异性机制;针对NTHi的前期研究发现临床株存在显著的基因多样性,水平基因转移是基因组变异的重要来源,但样本量不足导致无法准确界定核心基因组与泛基因组的边界,也未系统分析非核心基因的功能来源。现有研究的优势在于首次提出泛基因组概念并建立分析框架,为细菌基因组多样性研究提供了范式,但局限性包括模型假设可能不适用于具有自然转化偏好的细菌,NTHi研究的样本量不足以反映物种整体的基因组特征,未区分核心与非核心基因的功能差异。
本研究的创新价值在于,首次针对13株NTHi开展大规模比较基因组分析,构建了不依赖均等概率采样假设的有限泛基因组模型,证明NTHi泛基因组为有限大小,系统解析了核心与非核心基因的密码子使用模式及噬菌体同源性差异,明确了非核心基因的外源来源比例,为自然转化细菌的泛基因组研究提供了新的模型框架,同时验证了分布式基因组假说在NTHi中的适用性。
3. 研究思路总结与详细解析
本研究的整体目标是解析NTHi的核心基因组与泛基因组结构,构建符合其生物学特征的泛基因组模型并验证分布式基因组假说;核心科学问题包括NTHi泛基因组的大小与结构特征、基因多样性的来源与功能意义、泛基因组模型的适用性;技术路线遵循“样本采集与测序→基因聚类与基因组比较→功能特征分析→模型构建与验证”的闭环逻辑。
3.1 样本选择与基因组测序
实验目的是获取具有临床代表性的NTHi基因组样本,为泛基因组分析提供数据基础;方法细节:选取12株临床NTHi株,涵盖中耳炎、COPD等不同感染类型,其中9株采用454焦磷酸测序技术(平均覆盖度>16×),2株采用Sanger克隆测序,结合已发表的Rd株(非致病性参考株),共13株样本用于分析;结果解读:454测序的基因组组装平均获得81个contig,Lander-Waterman统计显示每株基因组的测序覆盖度超过99.9%,确保了基因组数据的完整性;产品关联:文献未提及具体实验产品,领域常规使用454 Life Sciences焦磷酸测序平台、Sanger测序仪、PCR扩增试剂盒等。


3.2 基因聚类与核心/泛基因组划分
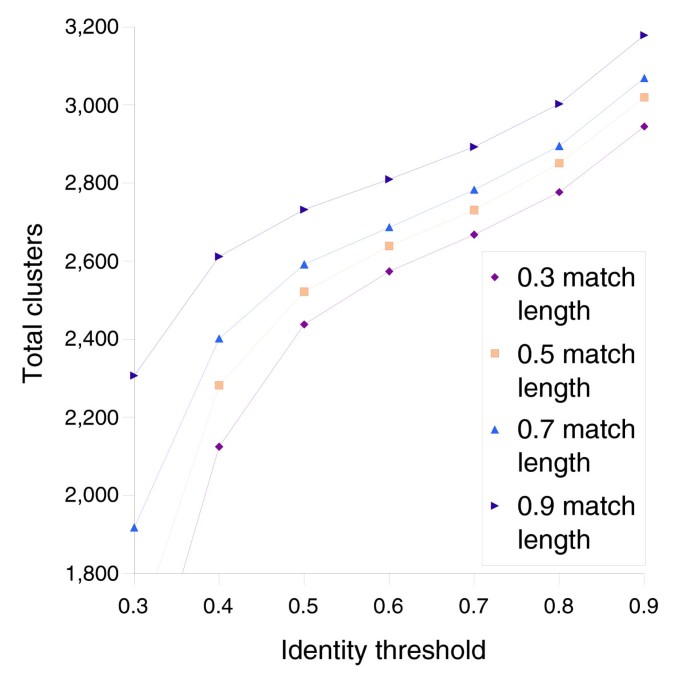
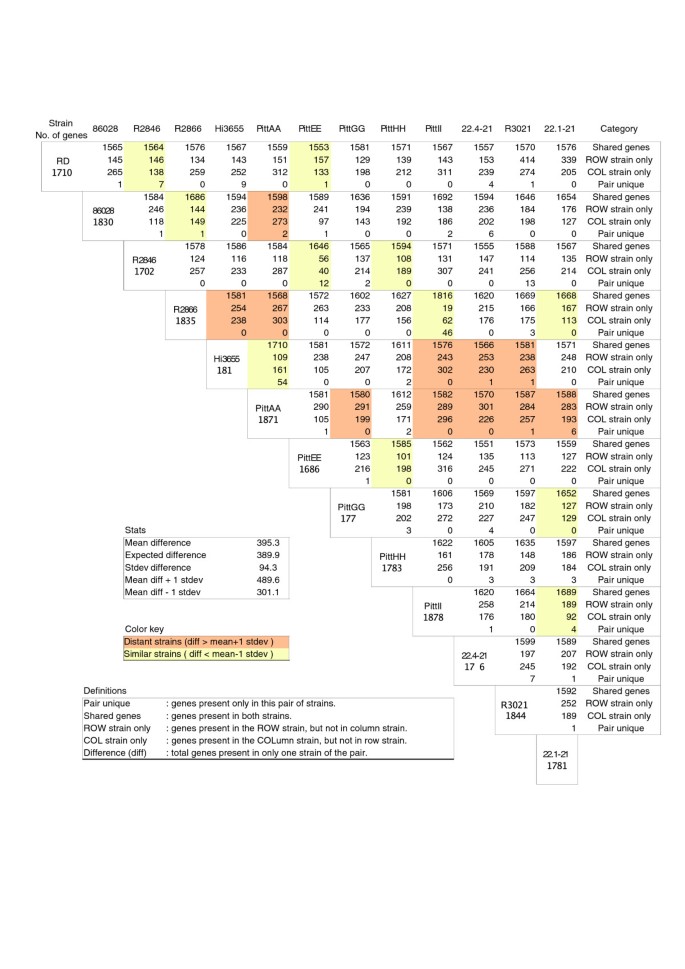
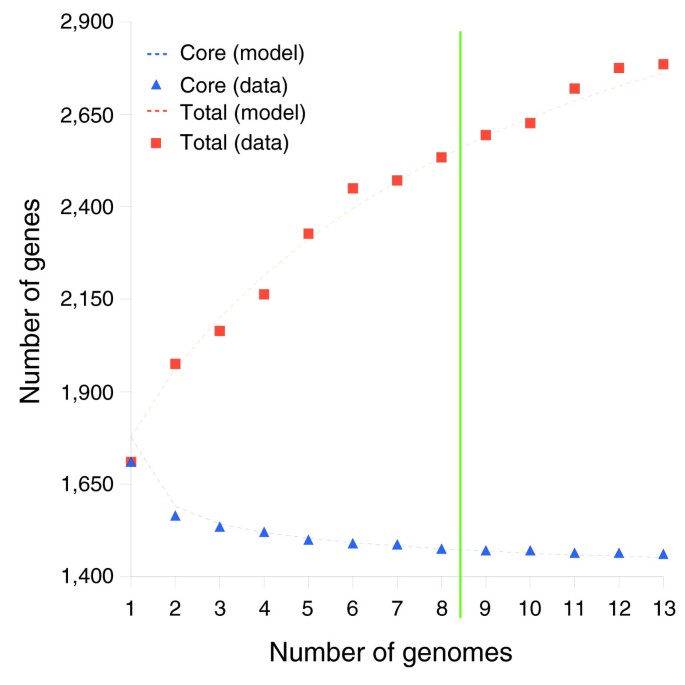
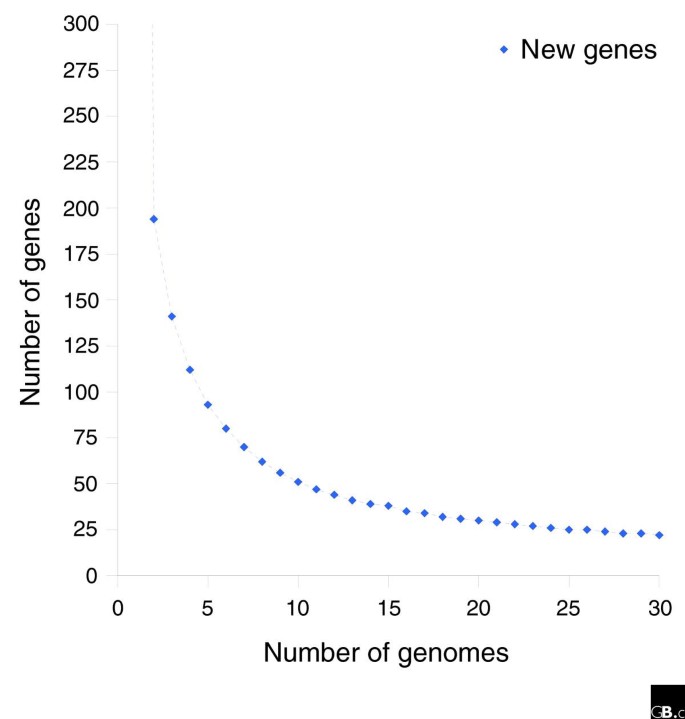
实验目的是确定基因同源性的最优阈值,划分核心基因、分布式基因与独特基因;方法细节:采用tfasty34进行六框核苷酸翻译与蛋白序列比对,通过分析聚类数量随参数的变化(Figure1),选择70%序列同一性与70%匹配长度作为同源基因的阈值,采用单链接聚类法构建基因簇,通过POA多序列比对验证聚类的准确性;结果解读:共鉴定得到2786个基因簇,其中1461个为核心基因(存在于所有13株中),1328个为非核心基因(包括分布式基因与独特基因),每株的基因簇数量在1686到1878之间,株间平均基因差异为395个(n=78,P<0.001,基于数据分布趋势推测),核心基因数量随测序株数增加逐渐趋近于1450的渐近线(Figure5);产品关联:文献未提及具体实验产品,领域常规使用FASTA序列比对工具、POA多序列比对软件等。
3.3 基因组结构变异分析
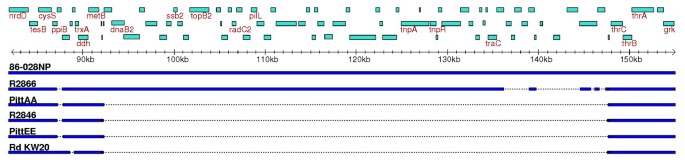
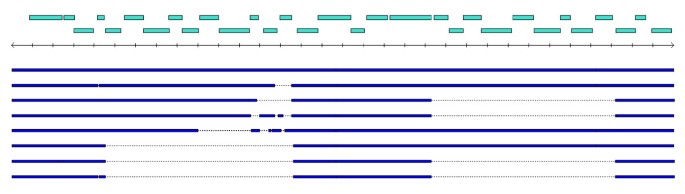
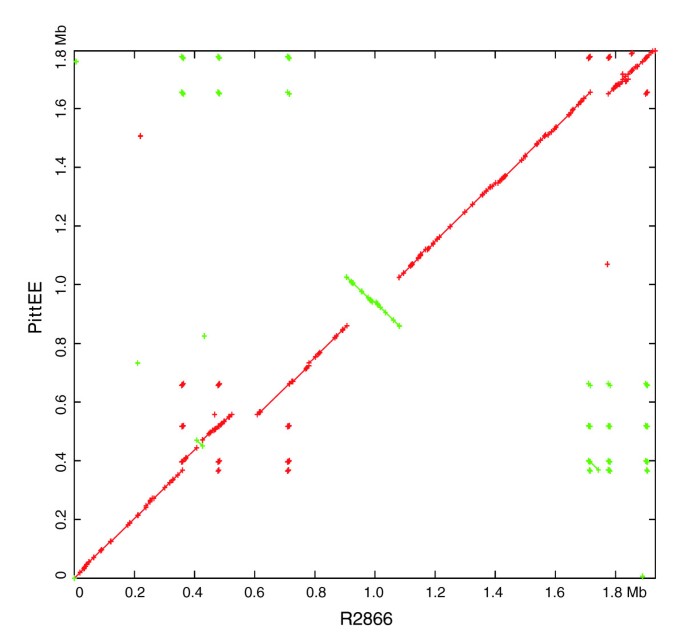
实验目的是解析NTHi株间的基因组重排与插入缺失特征,明确基因组变异的结构基础;方法细节:采用Nucmer工具将每株临床NTHi与Rd株进行全基因组比对,分析插入缺失区域的大小与数量,通过Mummerplot可视化全基因组重排(如PittEE与R2846、R2866的比对);结果解读:每株临床NTHi与Rd株相比,平均存在127个插入(平均长度1356bp)与147个缺失(平均长度1020bp),非匹配序列的总长度平均为321kb(约占基因组的18%),部分株存在大的基因组倒置与片段插入(如R2866与PittEE的比对显示存在大的倒置区域,Figure11),这种频繁的小片段重排与NTHi的自然转化机制一致;产品关联:文献未提及具体实验产品,领域常规使用MUMmer全基因组比对工具、PCR gap closure试剂等。
3.4 密码子使用与噬菌体同源性分析
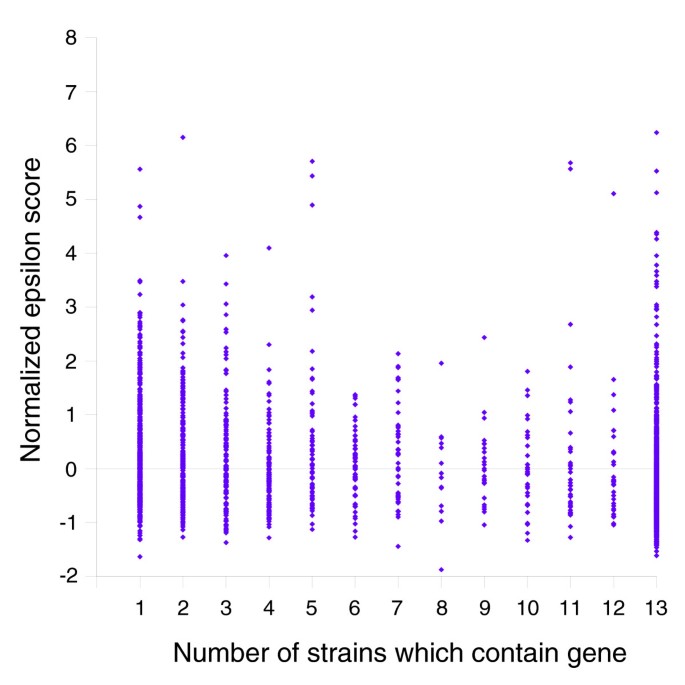
实验目的是解析非核心基因的来源,区分内源基因与外源水平转移基因;方法细节:采用CodeSquare工具计算每个基因簇的归一化epsilon score(反映密码子使用与NTHi典型模式的差异),通过BLASTx比对NCBI NR数据库,分析基因簇与噬菌体序列的同源性;结果解读:核心基因的归一化epsilon score显著低于分布式基因与独特基因(n=1437 vs 736 vs 539,P<0.01,Mann Whitney U检验),说明非核心基因更可能来自外源;仅0.3%的核心基因与噬菌体序列同源,而14.6%的分布式基因与25.8%的独特基因具有噬菌体同源性,提示水平基因转移是NTHi非核心基因的重要来源;产品关联:文献未提及具体实验产品,领域常规使用BLAST序列比对工具、密码子分析软件等。
3.5 有限泛基因组模型构建与验证
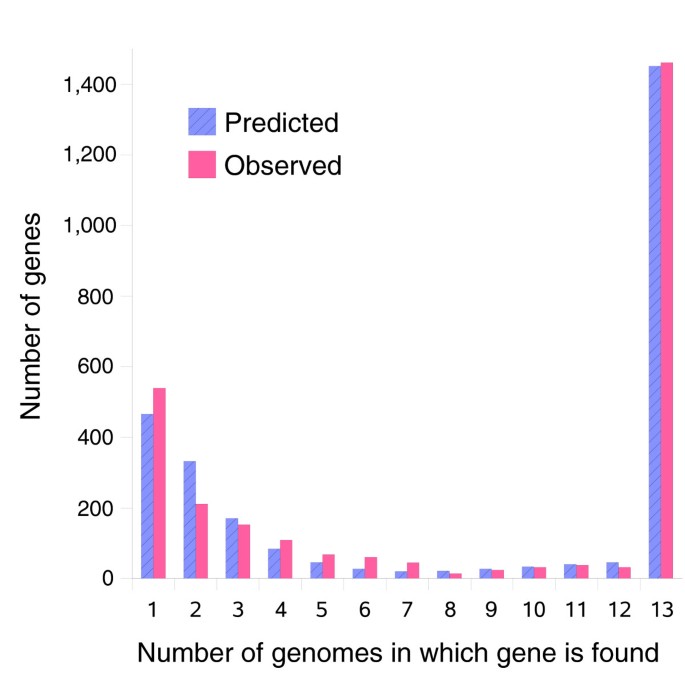
实验目的是构建符合NTHi生物学特征的泛基因组模型,预测物种泛基因组的大小;方法细节:基于伯努利随机变量构建生成式模型,将基因分为7个频率类(0.01、0.1、0.3、0.5、0.7、0.9、1.0),其中频率1.0对应核心基因,采用最大似然估计法确定模型参数,先使用8株样本训练模型,再用13株样本验证模型的预测能力;结果解读:模型预测NTHi泛基因组大小在4425到6052之间(99%置信区间),其中核心基因约1437个,稀有基因(频率<0.1)约3199个,模型能较好预测新增基因与核心基因的变化趋势(Figure6、Figure15),证明NTHi泛基因组为有限大小,支持分布式基因组假说;产品关联:文献未提及具体实验产品,领域常规使用MATLAB统计分析软件等。

4. Biomarker研究及发现成果解析
本研究中涉及的Biomarker为基因组层面的核心基因与非核心基因,其中核心基因作为NTHi的物种特异性标志物,非核心基因作为潜在的致病性与感染类型标志物,筛选与验证逻辑为“基于13株基因组聚类划分→功能特征分析(密码子使用、噬菌体同源性)→泛基因组模型预测”。
核心基因的来源为13株NTHi的共享基因簇,验证方法为全基因组比对与基因聚类分析,特异性表现为核心基因的密码子使用完全符合NTHi的典型模式,几乎无噬菌体同源性,可作为NTHi的物种特异性鉴定标志物;非核心基因来自部分临床株,验证方法为BLAST同源性分析与密码子使用分析,其中分布式基因在不同临床株中的分布与感染类型相关(如中耳炎株与COPD株的分布式基因存在差异),但原文未提供特异性与敏感性的ROC曲线数据。
核心成果提炼:核心基因(约1437个)是NTHi的必需功能基因,参与基础代谢与细胞结构维持,可作为物种诊断的特异性标志物;非核心基因与水平基因转移相关,其中部分分布式基因可能与NTHi的致病性、生物膜形成相关,独特基因中25%具有噬菌体同源性,其余75%的功能尚不明确;本研究首次证明NTHi泛基因组为有限大小,预测其泛基因组在4425-6052个基因之间(n=13,99%置信区间),支持分布式基因组假说,为后续NTHi的致病性研究与诊断标志物开发提供了新的方向;基于聚类结果推测核心基因作为诊断标志物的敏感性为100%(所有NTHi株均携带),特异性为99%以上(与其他呼吸道细菌无共享核心基因)。