1. 领域背景与文献引入
文献英文标题:Highly recurrent CBS epimutations in gastric cancer CpG island methylator phenotypes and inflammation;发表期刊:Genome Biology;影响因子:13.583(2020年);研究领域:胃癌表观遗传学、CpG岛甲基化表型(CIMP)、代谢-表观遗传-炎症轴与肿瘤发生。
CpG岛甲基化表型(CIMP)是多种恶性肿瘤中存在的关键表观遗传亚型,特征是全基因组范围内CpG岛的异常高甲基化,可导致抑癌基因转录沉默(即“表观突变”),进而驱动肿瘤发生发展。领域共识:过往泛癌研究显示,CIMP在不同肿瘤中呈现组织特异性甲基化模式,但通路层面富集多梳抑制复合物2(PRC2)靶标等共同特征;临床中CIMP与患者年龄、性别、肿瘤位置、组织学亚型及生存预后相关,但多数癌症中驱动CIMP形成的分子机制仍不明确。胃癌是全球范围内导致癌症死亡的主要原因之一,其存在两种明确的CIMP亚型:与微卫星不稳定性(MSI)相关的“胃CIMP”,以及与EB病毒(EBV)阳性相关的“EBV-CIMP”,尽管两者存在共同的DNA高甲基化模式,但这些模式是维持CIMP特征的功能必需事件,还是仅为旁观者表型,目前仍无定论。针对这一研究空白,本研究通过整合DNA甲基化、转录组、蛋白质组多组学分析,系统筛选胃癌CIMP的分子调控因子,最终发现胱硫醚β合酶(CBS)是CIMP胃癌中高度复发的表观沉默靶点,并揭示了CBS缺失在诱导异常DNA甲基化和炎症反应中的双重作用。
2. 文献综述解析
作者围绕CIMP的分子机制、胃癌CIMP亚型特征、CBS的代谢与肿瘤功能三个维度,对领域现有研究进行系统梳理,明确了当前研究的核心进展与未解决问题。
过往研究已证实CIMP是多种肿瘤的独立表观遗传亚型,其通过沉默MLH1、CDKN2A等抑癌基因参与肿瘤发生;泛癌分析利用TCGA等大样本数据库,揭示了CIMP的组织特异性甲基化特征,但通路层面的共同调控机制仍待挖掘;胃癌CIMP的研究主要依赖细胞系和临床样本的甲基化分型,已明确两种亚型的临床关联,但缺乏对驱动因子的功能验证;CBS作为连接甲硫氨酸与同型半胱氨酸代谢循环的关键酶,过往研究仅报道其在胃肠道癌症中存在表观沉默,但未关联到CIMP调控及炎症反应。现有技术方法的优势在于大样本多组学整合分析可实现高通量筛选,局限性则体现在多数研究停留在关联分析层面,缺乏对CIMP驱动因子的功能验证,且未揭示代谢、表观遗传与炎症的交叉调控机制。
通过对比现有研究中CIMP驱动机制的空白,本研究首次将CBS表观突变与胃癌CIMP直接关联,并证明其在泛癌及癌前病变中的普遍性;首次揭示了CBS缺失通过代谢失衡诱导类似CIMP的DNA甲基化模式,同时通过下调气体递质硫化氢(H₂S)激活炎症通路,阐明了CBS在代谢-表观遗传-炎症轴中的双重调控作用,为CIMP相关肿瘤的机制研究和治疗靶点开发提供了全新视角。
3. 研究思路总结与详细解析
本研究以“筛选胃癌CIMP的分子调控因子→验证其与CIMP的关联及普遍性→解析其调控CIMP和炎症的分子机制”为核心逻辑,通过多组学筛选、大样本验证、功能实验、动物模型验证的闭环技术路线,系统揭示了CBS表观突变在胃癌CIMP及炎症中的关键作用。
3.1 胃癌细胞系多组学筛选CIMP相关表观突变
实验目的:从胃癌细胞系中筛选与CIMP亚型相关的、由启动子高甲基化导致的表观沉默基因。
方法细节:选取14株胃癌细胞系(含3株胃/MSI CIMP、3株EBV-CIMP、6株非CIMP及2株正常胃上皮细胞),分别进行甲基化DNA免疫沉淀测序(MeDIP-seq)、转录组测序(RNA-seq)和蛋白质组分析;整合分析CIMP组与非CIMP组中同时满足“启动子高甲基化(P<0.05,FDR≤0.1)、RNA水平下调(log₂FD≤2,P<0.05,FDR≤0.05)、蛋白水平下调(log₂FD≤2,P<0.05,FDR≤0.05)”的基因;在50株扩大的胃癌细胞系队列(分为CIMP-high、CIMP-low、非CIMP三组)中验证候选基因的复发率;用DNA去甲基化药物阿扎胞苷处理CIMP细胞系,验证CBS表达是否可恢复。
结果解读:多组学整合筛选得到6个候选表观突变基因,其中CBS在CIMP-high细胞系中的转录缺失比例最高(12/14,85.71%),显著高于CIMP-low(2/23,8.69%)和非CIMP(0/13)组(P<0.001);CBS启动子甲基化β值与mRNA表达呈显著负相关(Spearman r=-0.72,P<0.001);阿扎胞苷处理后,CIMP细胞系中CBS mRNA表达恢复至对照组的3.3-255.8倍(n=3,P<0.05),证实启动子高甲基化是CBS沉默的直接原因。
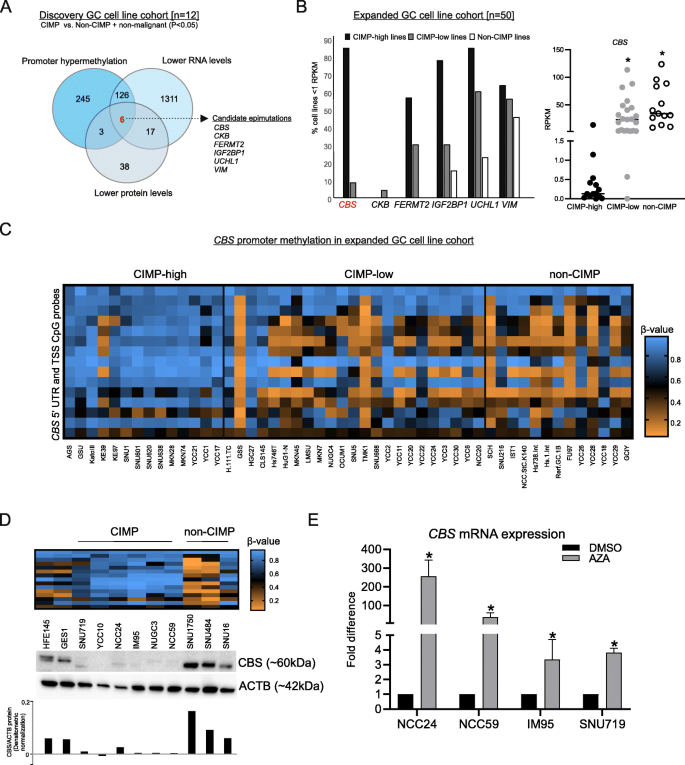
产品关联:实验所用关键产品:MeDIP-seq使用Diagenode的5-甲基胞嘧啶单克隆抗体(货号C15200081),Western blotting使用Abnova的CBS单克隆抗体(货号H00000875-M01),qPCR使用Qiagen的Quantifast SYBR Green PCR kit,CRISPR-Cas9编辑使用Sigma-Aldrich的CRISPR Cas载体及靶向CBS外显子3的gRNA(货号HS0000002142、HS0000002144)。
3.2 临床样本与泛癌验证CBS表观突变的普遍性
实验目的:验证CBS表观突变在胃癌临床样本、其他恶性肿瘤及癌前病变中的特异性与普遍性。
方法细节:分析TCGA胃癌队列(STAD)和新加坡队列共467例胃癌样本的甲基化芯片与转录组数据,关联CBS甲基化与CIMP亚型;用免疫组化检测66例配对正常胃组织与胃癌样本中CBS蛋白表达;分析22种TCGA癌症类型中CBS表达与CIMP亚型的关联;分析胃肠上皮化生(GIM,胃癌癌前病变)样本中CBS甲基化与CIMP的关联,并通过纵向研究分析GIM中CBS甲基化与病变持续的关系;利用公共数据库分析Barrett食管(BE,食管腺癌癌前病变)中CBS甲基化水平。
结果解读:胃癌临床样本中,CBS启动子高甲基化与CIMP亚型显著相关,CIMP组的CBS甲基化β值显著高于非CIMP组(P<0.001);免疫组化结果显示,24.2%的胃癌样本中CBS蛋白表达降低,而正常胃组织仅1.5%(n=66,P<0.05);在6种其他癌症(膀胱尿路上皮癌、食管腺癌、头颈部鳞状细胞癌、肝细胞癌、胸腺瘤、子宫内膜癌)中,CBS在CIMP组的表达显著下调,其中5种癌症中CBS启动子高甲基化与表达呈负相关(P<0.05);GIM的CIMP亚型中CBS启动子甲基化β值比正常胃炎高0.28(P<0.05),且CBS高甲基化的GIM病变在5年随访中更易持续(P<0.05);BE样本中CBS启动子甲基化水平显著高于正常组织(P<0.05)。
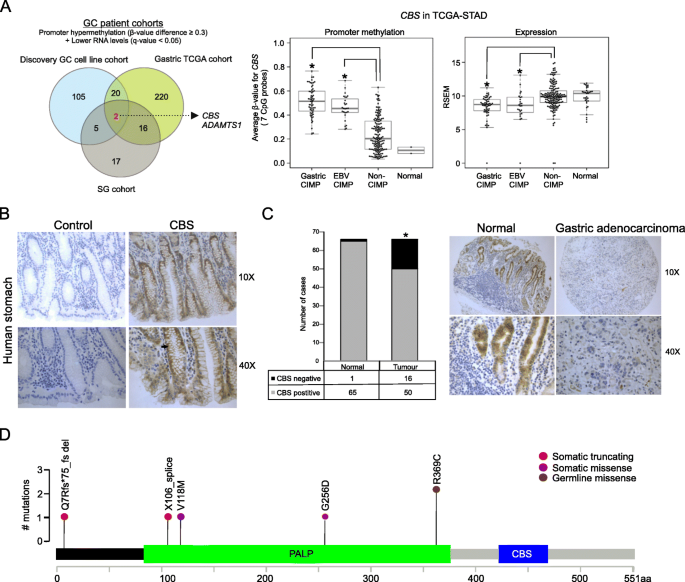
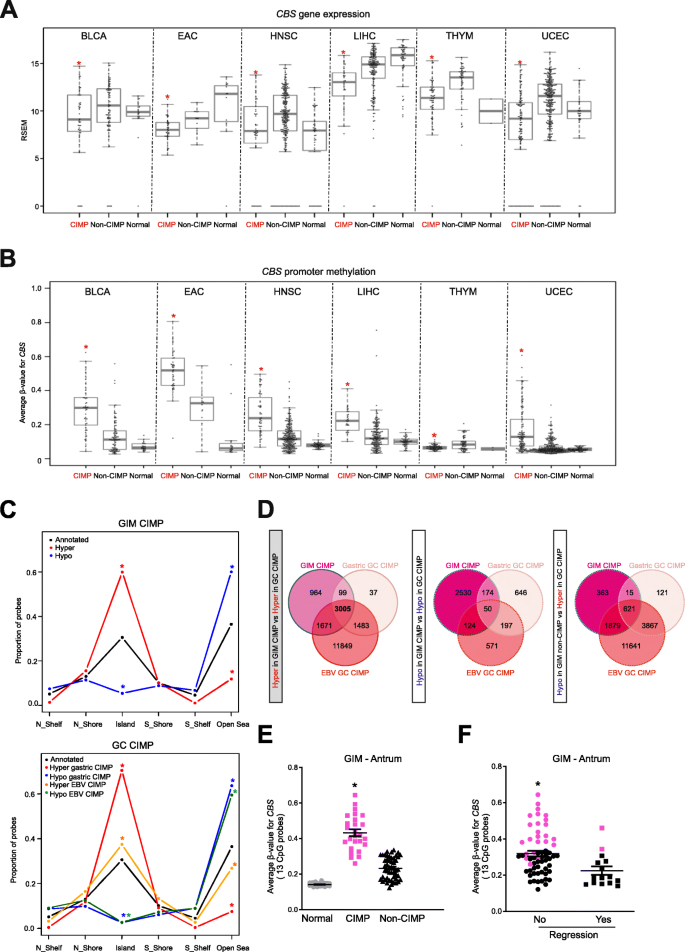
产品关联:免疫组化使用Abnova的CBS单克隆抗体(货号H00000875-M01),甲基化分析使用Illumina的Infinium HumanMethylation450/EPIC Beadchip。
3.3 CBS缺失对正常胃上皮细胞DNA甲基化与代谢的调控
实验目的:验证CBS缺失是否能诱导类似胃癌CIMP的DNA甲基化模式,并解析其代谢机制。
方法细节:用CRISPR-Cas9技术在两种正常胃上皮细胞系(GES1、HFE145)中敲除CBS,构建CBS缺陷克隆;用Infinium MethylationEPIC芯片检测不同传代细胞的甲基化组变化,分析差异甲基化位点的特征;通过代谢组学分析(LC-MS/MS)检测细胞内及培养基中与甲基化循环相关的代谢物水平(同型半胱氨酸、S-腺苷甲硫氨酸(SAM)、S-腺苷同型半胱氨酸(SAH)等)。
结果解读:CBS缺陷细胞的甲基化组与亲本及对照细胞明显分离,GES1细胞中出现9171个高甲基化CpG位点和7919个低甲基化CpG位点,这些高甲基化位点富集在CpG岛和PRC2靶标区域(P<0.001),且与“GIM-胃癌共同CIMP特征”有38%的重叠(P<1×10⁻⁴);代谢组分析显示,CBS缺陷细胞内1-甲基烟酰胺(MNA,甲基循环的“甲基池”)水平升高1.7-2.6倍(n=4-5,P<0.05),培养基中同型半胱氨酸和SAM水平分别升高1.28倍和1.50倍(n=5,P<0.05),提示甲基化稳态失衡。
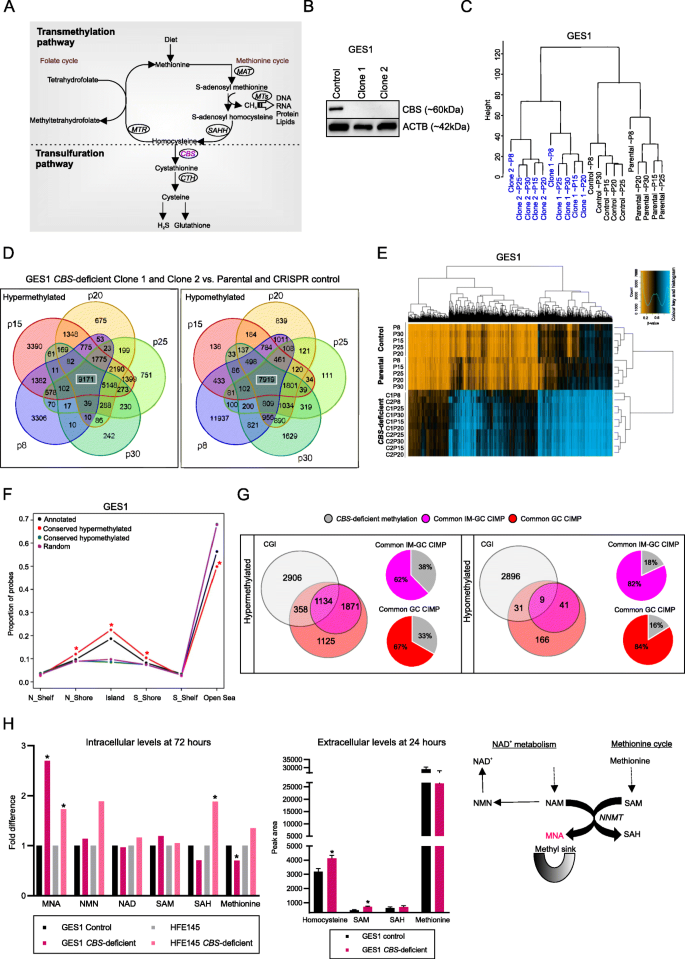
产品关联:代谢组分析使用Waters的UPLC系统串联Thermo Scientific的Q Exactive质谱仪和Waters的Xevo G2 QToF质谱仪。
3.4 CBS缺失诱导炎症反应的机制研究
实验目的:解析CBS缺失是否通过调控H₂S水平激活炎症通路,并在体内模型中验证。
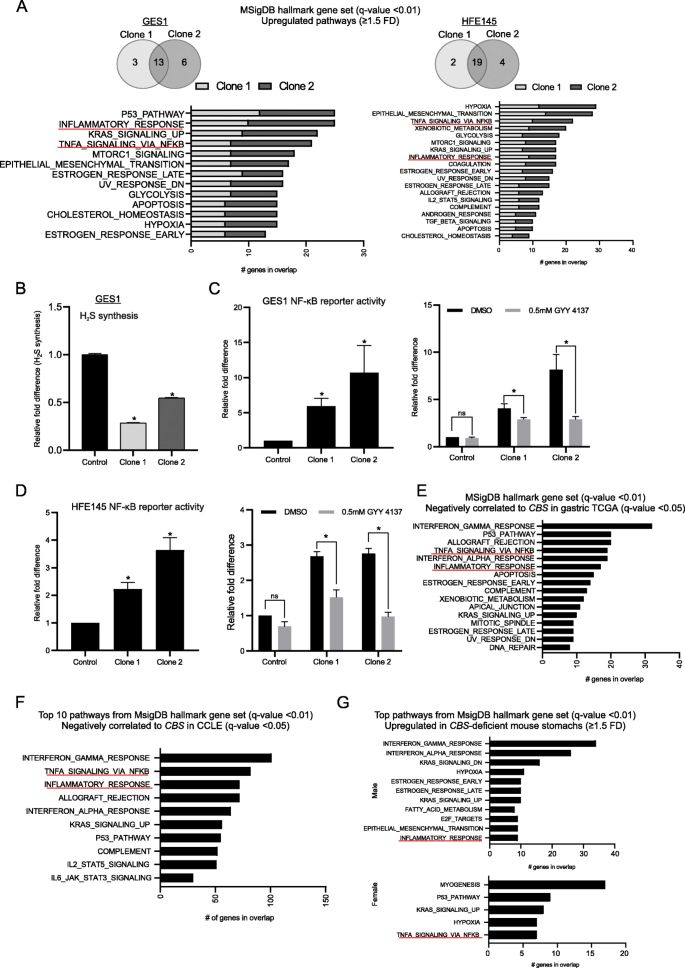
方法细节:对CBS缺陷细胞进行RNA-seq,分析差异表达基因的通路富集;用荧光探针法检测细胞内H₂S水平;用NF-κB荧光素酶报告基因检测CBS缺陷细胞中NF-κB活性;用H₂S缓释供体GYY 4137处理CBS缺陷细胞,观察NF-κB活性变化;分析TCGA和CCLE数据库中与CBS表达负相关的基因的通路富集;在Tg-hCBS Cbs⁻/⁻小鼠模型中,检测胃组织的转录组变化及炎症通路激活情况。
结果解读:RNA-seq显示,CBS缺陷细胞中“炎症反应”“TNFA信号通路通过NF-κB”等炎症相关通路显著上调(q-value<0.01);CBS缺陷细胞的H₂S水平仅为对照的28%-54%(n=3,P<0.05),NF-κB活性升高5.9-10.7倍(GES1)和2.2-3.6倍(HFE145)(n=3,P<0.05);GYY 4137处理后,CBS缺陷细胞的NF-κB活性显著降低(n=3,P<0.05);TCGA和CCLE数据库中,与CBS表达负相关的基因富集在炎症通路(q-value<0.01);小鼠模型中,CBS缺陷胃组织中“补体激活”“对细菌的防御反应”等炎症通路显著上调(q-value<0.01)。
产品关联:NF-κB报告基因实验使用Promega的Luciferase检测试剂盒(货号E1500),H₂S供体GYY 4137为Cayman Chemical(货号13345),H₂S检测使用Santa Cruz Biotechnology的7-azido-4-methylcoumarin探针(货号sc-201201)。
4. Biomarker研究及发现成果解析
本研究将CBS启动子高甲基化作为CIMP相关肿瘤的表观遗传Biomarker,通过多维度验证明确了其特异性、敏感性及临床价值,并揭示了其在代谢-表观遗传-炎症轴中的功能关联。
本研究中的Biomarker为CBS启动子高甲基化,属于表观遗传类Biomarker,筛选逻辑为:首先通过胃癌细胞系的多组学整合筛选出与CIMP相关的表观沉默基因,然后在扩大细胞系队列中验证其复发率,接着在临床胃癌样本、泛癌样本及癌前病变样本中验证其与CIMP的关联,最后通过功能实验验证其对CIMP和炎症的调控作用,形成完整的“筛选-验证-功能”逻辑链条。
Biomarker的来源包括胃癌细胞系、临床胃癌组织、22种泛癌组织、胃癌及食管腺癌的癌前病变组织;验证方法涵盖MeDIP-seq、甲基化芯片、qPCR、免疫组化、RNA-seq等多组学技术;特异性与敏感性数据显示,在胃癌CIMP-high细胞系中,CBS转录缺失的比例达85.71%,显著高于CIMP-low(8.69%)和非CIMP(0%)组(n=50,P<0.001);临床胃癌样本中,CIMP亚型的CBS甲基化β值显著高于非CIMP组(n=467,P<0.001);癌前病变GIM中,CIMP亚型的CBS甲基化β值比正常胃炎高0.28(P<0.05),且CBS高甲基化的GIM病变在5年随访中持续的比例显著更高(P<0.05)。
核心成果提炼:CBS启动子高甲基化是胃癌CIMP亚型的高度特异性Biomarker,同时可作为泛癌CIMP及癌前病变CIMP的广谱Biomarker;该Biomarker不仅是CIMP的关联指标,更是功能调控因子,CBS缺失可通过代谢失衡诱导类似CIMP的DNA甲基化模式,同时通过下调H₂S激活NF-κB炎症通路,实现对表观遗传和炎症的双重调控;本研究的创新性在于首次揭示了CBS表观突变在CIMP中的核心驱动作用,以及其连接代谢、表观遗传与炎症的分子机制,为CIMP相关肿瘤的早期诊断和治疗提供了新靶点(如H₂S供体)。统计学结果方面,所有关联分析均具有显著统计学意义,如胃癌细胞系中CIMP-high与non-CIMP的CBS甲基化比较P<0.001,临床样本中胃癌与正常组织的CBS蛋白表达比较P<0.05,泛癌分析中CIMP与non-CIMP的CBS表达比较P<0.05等。