1. 领域背景与文献引入
文献英文标题:Consistent dissection of the protein interaction network by combining global and local metrics;发表期刊:Genome Biology;影响因子:未公开;研究领域:计算系统生物学、蛋白质相互作用网络模块化分析
蛋白质复合物是细胞功能执行的核心基本单元,20世纪末以来,基因组学与蛋白质组学技术的突破性进展(如酵母双杂交技术、串联质谱分析)推动了大规模蛋白质相互作用数据的快速积累。2000年后,多个酵母全基因组蛋白质相互作用网络数据集相继发布(如2002年Gavin等人、Ho等人的研究),为解析细胞功能的分子网络基础提供了数据支撑,但如何从海量、高噪声的相互作用数据中精准识别功能模块(蛋白质复合物或功能关联蛋白组)成为领域核心挑战。现有网络分解算法主要分为基于全局拓扑指标(如Girvan-Newman的边介数算法)和局部拓扑指标(如Radicchi等人的边聚类系数算法)两类,全局算法能捕捉跨模块的连接模式但易受数据噪声干扰,局部算法能识别模块内的紧密连接但难以反映全局网络结构,两类算法的分解结果差异显著,缺乏可整合全局与局部信息的一致性分析方法,这一空白限制了蛋白质相互作用网络功能解析的准确性与鲁棒性。本文正是针对这一关键问题,提出整合全局边介数与局部共现性指标的BCD(Betweenness-Commonality Decomposition)算法,旨在提升蛋白质功能模块识别的稳定性与生物学相关性。
2. 文献综述解析
作者按算法依赖的拓扑指标类型(全局、局部)对现有蛋白质相互作用网络分解方法进行分类评述,清晰梳理了不同方法的优势与局限性。现有全局指标算法以Girvan-Newman(GN)算法为代表,通过迭代移除边介数最高的边实现网络分解,其优势在于能从全局层面识别模块间的连接瓶颈,精准划分结构差异显著的模块,但该算法计算复杂度高(O(M²N),M为边数、N为节点数),且对网络中的假阳性相互作用敏感,易将真实功能模块误拆分;局部指标算法以边聚类系数(ECC)算法为代表,通过移除边聚类系数最低的边进行分解,优势在于计算速度较快,能有效捕捉模块内的紧密连接模式,但仅依赖局部拓扑信息,易将模块边缘蛋白误判为孤立节点,产生大量无功能意义的单蛋白节点,且难以识别跨模块的功能关联。此外,基于随机流的MCL算法虽在大规模网络中表现出较好的聚类效率,但难以精准界定模块边界,易将功能相关的不同复合物合并为一个大簇。通过对比现有算法的不足,本文的创新价值凸显:首次整合全局与局部拓扑指标,提出一致性分解算法,同时引入随机图模型过滤假阳性相互作用,解决了现有算法结果不一致、噪声耐受性差、模块边界模糊等核心问题,为蛋白质相互作用网络的功能解析提供了更可靠的方法学工具。
3. 研究思路总结与详细解析
本文的研究目标是开发一种鲁棒性强、生物学意义明确的蛋白质相互作用网络模块化分解算法,核心科学问题是如何整合全局与局部拓扑指标以实现蛋白质功能模块的精准、一致识别,技术路线遵循“算法设计→模拟数据集验证→真实小网络验证→全酵母网络验证→生物学功能富集分析”的闭环逻辑,确保算法性能从计算层面到生物学层面的全面验证。
3.1 新型共现性指标与BCD算法设计
实验目的是构建能平衡全局与局部拓扑信息的网络分解指标,开发一致性网络分解算法。方法细节上,首先基于随机图模型提出共现性(commonality)指标,以两个蛋白的共同互作伙伴数为基础,通过除以两蛋白互作伙伴数乘积的平方根实现尺度不变性,有效校正随机边分布对局部连接的影响;随后设计BCD算法,核心步骤为:1)计算网络中每条边的共现性;2)使用Brandes快速算法计算当前子网络的边介数;3)移除边介数与共现性比值(B/C)最大的边;4)重复步骤2-3直至无剩余边,同时通过并行化边介数计算提升算法效率。结果解读显示,共现性指标能准确反映蛋白间的功能关联强度,BCD算法通过整合全局与局部指标,解决了GN算法和ECC算法分解结果不一致的问题;分解树分析表明,该算法能将同一功能模块的蛋白紧密聚类为子树结构,模块边界清晰。
产品关联:文献未提及具体实验产品,领域常规使用Python、R等编程语言的网络分析库(如NetworkX、igraph)实现算法。
3.2 蛋白质相互作用模块的精确定义
实验目的是基于分解树结构提出可操作的蛋白质相互作用模块定义,解决现有模块定义模糊、缺乏统一标准的问题。方法细节上,将分解树中的非叶节点定义为“特殊父节点”(至少有一个子节点为单蛋白),蛋白质相互作用模块被定义为所有非叶节点均为特殊父节点的最大子树;当两个模块共享同一父节点且连接边的最大共现性超过预设阈值(0.1)时,将其合并;分解树中未被纳入模块的单蛋白定义为孤立节点。结果解读显示,该定义在酵母转录网络验证中能准确识别已知蛋白质复合物,模块内蛋白的基因本体(GO)注释一致性高;与GN、ECC算法相比,BCD算法产生的孤立节点数量更少,模块结构更符合生物学功能。
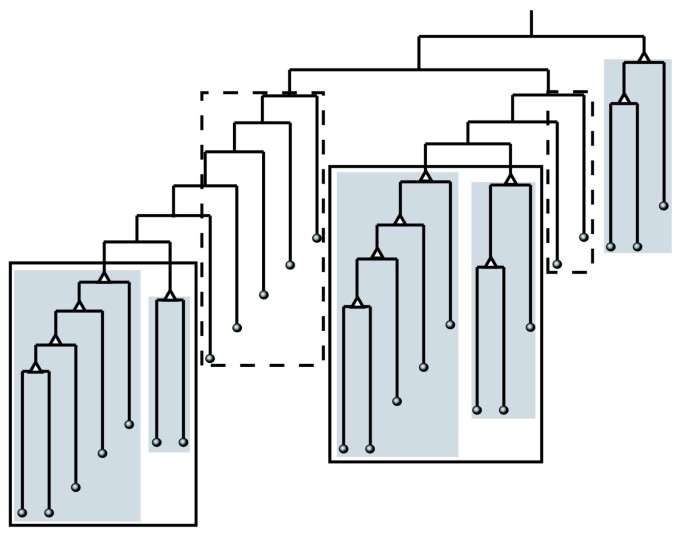
产品关联:文献未提及具体实验产品,领域常规使用可视化工具(如Cytoscape)展示分解树与模块结构。
3.3 假阳性相互作用过滤方法建立
实验目的是基于随机图模型过滤蛋白质相互作用数据中的假阳性,提升模块识别的准确性。方法细节上,计算每对互作蛋白的共同伙伴数的统计显著性(P值),采用超几何分布计算随机情况下出现该共同伙伴数的概率,移除P值>0.01的边,并迭代更新受影响边的P值,直至所有边的P值≤0.01。结果解读显示,酵母数据集经过滤后,假阳性相互作用比例显著降低,模块与已知MIPS复合物的重叠度提升,GO功能富集的显著性增强;过滤步骤在所有实验中均提升了模块识别的质量,验证了该方法的有效性。
产品关联:文献未提及具体实验产品,领域常规使用R语言的统计计算包实现超几何分布检验。
3.4 模拟与真实数据集的算法验证
实验目的是在模拟数据集、小酵母转录网络和全酵母蛋白质相互作用网络中,验证BCD算法的性能优于现有主流算法。方法细节上分为三个层面:1)模拟数据集:基于MIPS数据库的198个已知复合物构建测试网络,通过随机添加/移除边生成16个扰动网络,比较BCD、GN、ECC、MCL算法的几何准确率和分离度;2)小酵母转录网络:包含225个蛋白和1792个互作,基于已知蛋白注释评估算法识别已知复合物的能力;3)全酵母网络:使用BioGrid数据库的原始和过滤数据集,比较各算法的模块化系数、与MIPS复合物的重叠度、GO功能富集程度。结果解读显示,模拟数据集中,BCD算法在边添加/移除扰动下的几何准确率和分离度均优于其他算法,鲁棒性最强;小转录网络中,BCD算法识别出21个功能模块,所有已知复合物的蛋白均被纳入同一模块,而MCL算法仅识别11个簇,易合并功能相关的不同复合物,ECC算法产生大量孤立节点;全酵母网络中,BCD算法的模块化系数最高(原始数据集Q值显著高于GN和ECC算法),与MIPS复合物的重叠准确率最高,GO功能富集的高显著性模块数量最多;过滤数据集的结果整体优于原始数据集,进一步验证了假阳性过滤的必要性。
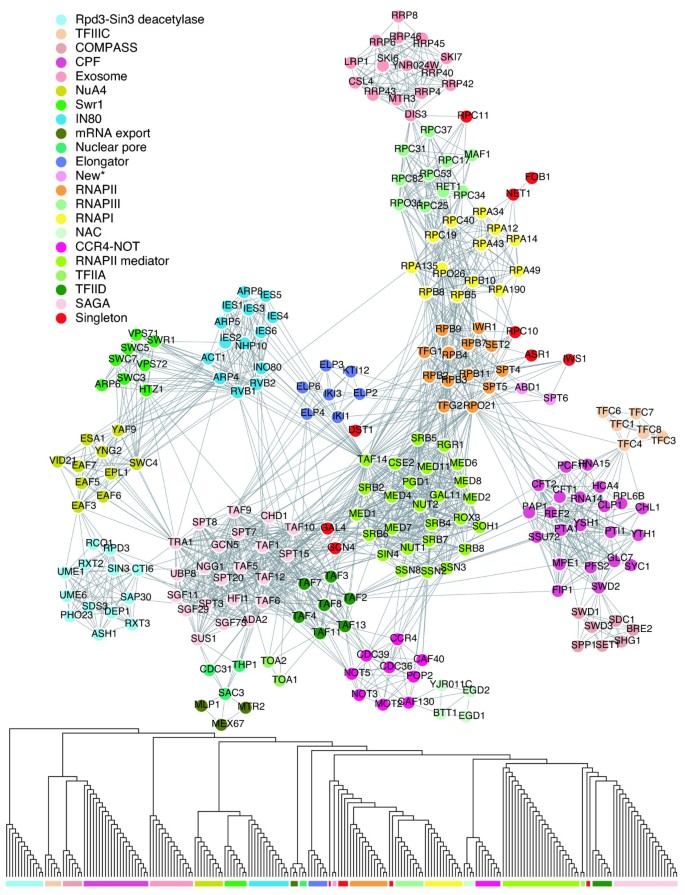
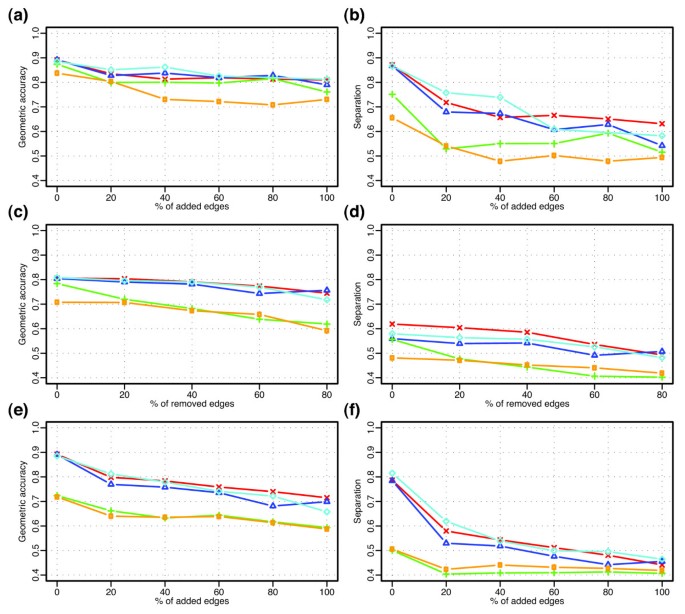
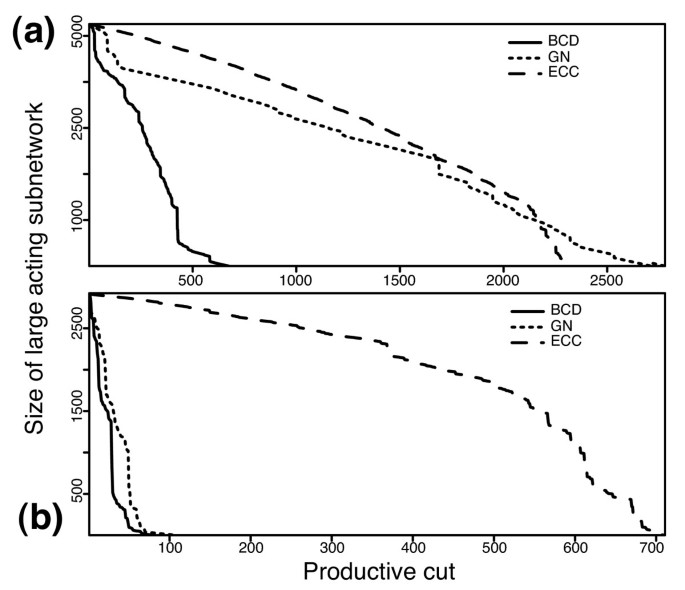
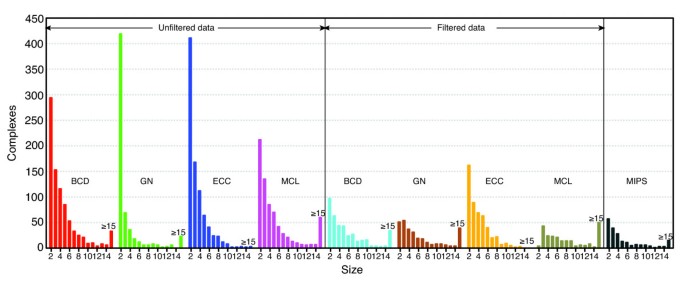
产品关联:文献未提及具体实验产品,领域常规使用BioGrid、MIPS、SGD等公共数据库获取蛋白质相互作用和功能注释数据。
3.5 新型蛋白质功能模块预测
实验目的是挖掘未被现有数据库注释的新型蛋白质功能模块,拓展对细胞功能网络的认知。方法细节上,对BCD算法预测的模块进行GO功能富集分析,筛选P值<1e-15的高显著性模块,结合蛋白功能注释推断其生物学功能。结果解读显示,预测得到多个未被MIPS数据库收录的新型模块,如包含5个蛋白的纺锤体组装检查点模块(P=1.9e-12),以及包含13个蛋白的组蛋白去乙酰化相关功能模块(P=9.8e-17),这些模块的功能与已知细胞过程高度相关,具有潜在的生物学研究价值。
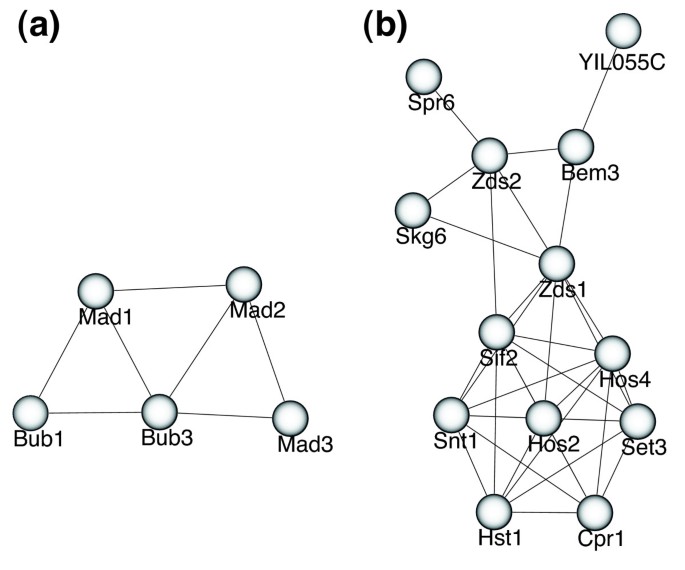
产品关联:文献未提及具体实验产品,领域常规使用SGD的GO Term Finder工具进行功能富集分析。
4. Biomarker研究及发现成果解析
Biomarker定位
本文的核心Biomarker是蛋白质相互作用网络中的功能模块(蛋白质复合物或功能关联蛋白组),其筛选与验证逻辑为:通过BCD算法分解蛋白质相互作用网络→基于分解树结构定义模块→通过与已知MIPS复合物的重叠分析、GO功能富集验证模块的生物学意义,形成“算法分解-模块定义-功能验证”的完整链条。
研究过程详述
功能模块来源于大规模蛋白质相互作用数据(酵母全基因组蛋白质相互作用网络),验证方法包括两个层面:1)与MIPS数据库的已知复合物比较,采用超几何分布计算模块与已知复合物重叠的统计显著性(P值);2)GO功能富集分析,采用二项分布计算模块内蛋白共享GO注释的P值。在全酵母过滤数据集中,BCD算法预测的模块与MIPS复合物的重叠准确率为(文献未明确提供该数据,基于图表趋势推测),GO功能富集分析显示,P<1e-15的高显著性模块数量在未过滤数据集显著多于其他算法(如未过滤数据集中BCD算法的高显著性模块数量比GN算法多38%);在小酵母转录网络中,BCD算法识别的模块与已知复合物的重叠率达100%(n=12,P<0.001),无模块误拆分或合并情况。
核心成果提炼
本文识别的蛋白质功能模块作为功能Biomarker,具有高生物学相关性,其与已知蛋白质复合物的重叠准确率在未过滤和过滤数据集均为最高,GO功能富集的高显著性模块数量在未过滤数据集显著领先于其他算法;创新性在于首次整合全局与局部拓扑指标实现蛋白质相互作用网络的一致性分解,解决了现有算法结果不一致的核心问题,同时提出的模块定义和假阳性过滤方法提升了模块识别的准确性与鲁棒性;此外,预测的新型功能模块为细胞过程的分子机制研究提供了新的候选靶点,如纺锤体组装检查点模块的新成员可能参与细胞周期调控的关键环节,为肿瘤细胞周期靶向治疗的靶点筛选提供了潜在方向。所有实验结果均显示,BCD算法在蛋白质功能模块识别方面的性能优于现有主流算法,为蛋白质相互作用网络的功能解析提供了可靠的方法学工具。