1. 领域背景与文献引入
文献英文标题:Mapping MAVE data for use in human genomics applications;发表期刊:Genome Biology;影响因子:未明确提供;研究领域:人类基因组学、功能基因组学、临床变异解读
高通量测序技术在临床基因组学中的应用持续增长,但其带来的变异解读缺口也日益凸显——ClinVar数据库中约一半的 curated变异被归类为“意义未明变异(variant of uncertain significance, VUS)”,缺乏足够的功能证据支持其致病性或良性判定。领域共识:计算预测工具虽已取得进展,但无法替代实验获得的功能数据,而多重变异效应分析(Multiplexed Assays of Variant Effect, MAVE)技术作为高通量功能评估手段,能同时检测数千个变异的功能效应,为变异解读提供关键实验证据。MaveDB作为多重变异效应分析数据的集中存储库,已积累了超过2000个实验数据集,但当前多重变异效应分析数据通常基于实验设计的特定序列生成,无法直接对应人类参考基因组,导致临床和研究中难以与其他基因组学数据整合使用。针对这一核心问题,本研究旨在建立多重变异效应分析数据与人类参考基因组的精准映射流程,将这些功能数据整合到常用的基因组学工具中,提升数据的可及性与利用率,辅助临床变异解读与基础研究。
2. 文献综述解析
本文的背景部分系统梳理了领域内的研究现状,作者按“临床测序挑战-功能评估技术-数据资源瓶颈”的维度分类评述现有研究。现有研究已证实多重变异效应分析技术在变异功能评估中的核心优势,能产生大规模、系统性的变异功能数据,MaveDB的建立实现了这些数据的集中存储与共享,为领域提供了宝贵的功能证据资源。但现有研究存在明显局限性:多重变异效应分析数据多基于实验设计的特定序列,与人类参考基因组缺乏标准化映射,导致数据无法直接对接临床常用的基因组学分析平台,难以与临床变异数据、蛋白结构数据等整合使用;同时,此前缺乏针对大规模多重变异效应分析数据的自动化映射流程,无法满足领域对这类数据规模化应用的需求。本文的创新价值在于,首次针对MaveDB中所有人类相关的多重变异效应分析数据集开展标准化映射,解决了实验序列与参考基因组的对接难题,并通过多平台整合实现了多重变异效应分析数据的广泛应用,填补了多重变异效应分析数据从实验存储到临床实用的关键空白。
3. 研究思路总结与详细解析
本文的研究目标是建立多重变异效应分析数据与人类GRCh38参考基因组的精准映射流程,同时保留实验数据的溯源信息,并将映射后的数据整合到主流人类基因组学工具中;核心科学问题是如何在维持实验数据完整性的前提下,实现基于实验特定序列的多重变异效应分析变异到人类参考基因组的标准化转换;技术路线遵循“数据筛选-序列比对-变异映射-平台整合-应用验证”的闭环逻辑。
3.1 MaveDB人类数据集筛选与元数据提取
本环节的核心目标是从MaveDB中筛选出针对人类序列的多重变异效应分析数据集,并提取关键元数据为后续映射提供基础。研究人员从最新版本的MaveDB中筛选出1064个以人类序列为靶标的得分集,通过API接口提取每个数据集的靶序列、序列类型(蛋白或核苷酸)、靶基因名称、UniProt ID等核心元数据。结果显示,这些数据集共包含超过900万个独立变异,其中1023个数据集针对蛋白编码基因,41个针对调控及其他非编码元件;582个数据集在氨基酸水平描述变异,482个在核苷酸水平描述变异。文献未提及具体实验产品,领域常规使用Python脚本、REST API接口类工具进行元数据提取与整理。
3.2 MAVE靶序列与人类参考基因组比对
本环节的核心目标是将多重变异效应分析实验靶序列与人类GRCh38参考基因组进行比对,确定同源区域及对应的标准转录本。研究人员使用BLAT工具将每个多重变异效应分析靶序列比对到GRCh38基因组,随后通过计算推断与靶序列兼容的RefSeq转录本,优先选择MANE Select转录本(临床变异报告的标准转录本),并计算多重变异效应分析序列在参考蛋白序列中的偏移量以精确定位变异位置。结果显示,1064个数据集中有1057个成功完成比对与转录本选择,仅7个数据集映射失败(6个无法确定对应的RefSeq转录本,1个靶序列无法与参考基因组比对)。比对过程中发现,部分多重变异效应分析序列因实验设计需求(如密码子优化、包含非同源实验元件)与参考序列存在差异,这些差异被完整记录以保留数据溯源。
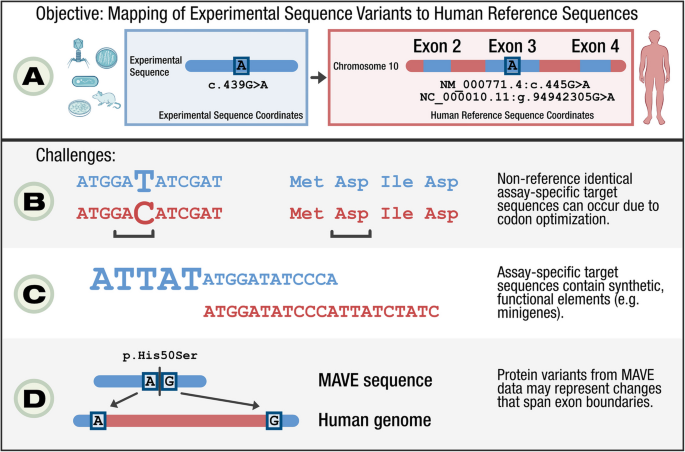
文献未提及具体实验产品,领域常规使用BLAT、SeqRepo、UTA等生物信息学工具进行序列比对与转录本选择。
3.3 基于GA4GH VRS标准的变异映射生成
本环节的核心目标是将多重变异效应分析变异转换为符合GA4GH变异表示规范(Variation Representation Specification, VRS)的标准化对象,建立原始实验变异与参考基因组变异的映射对,同时保留数据溯源信息。研究人员将每个多重变异效应分析变异转换为VRS等位基因对象,计算序列摘要并注册到SeqRepo数据库,对变异进行规范化处理以确保表示的一致性;同时为每个映射对添加HGVS命名,提升与下游工具的互操作性。结果显示,共生成6158451个一致的变异映射对,对应1048823个独特的原始实验变异和1018091个独特的参考基因组变异;其中899个数据集的靶序列与参考序列存在差异,通过映射首次实现了820427个独特参考变异的功能数据访问。每个映射对包含原始变异(基于实验序列)和映射后变异(基于参考基因组)的完整信息,并分配唯一标识符以确保数据溯源的可追踪性。文献未提及具体实验产品,领域常规使用VRS-Python、Cool-Seq-Tool等工具进行变异标准化处理。
3.4 映射后MAVE数据的多平台整合与验证
本环节的核心目标是将映射后的多重变异效应分析数据整合到常用的人类基因组学研究与临床变异解读工具中,验证数据的可及性与实用性。研究人员针对不同平台开发定制化整合流程:在Genomics 2 Proteins(G2P)Portal中整合706个得分集,实现多重变异效应分析数据在蛋白序列和三维结构上的可视化;在UCSC Genome Browser中创建自定义track hub,以热图形式展示变异的功能得分;为Ensembl Variant Effect Predictor(VEP)开发专用插件,实现变异注释时自动调用多重变异效应分析数据;在DECIPHER平台的变异页面添加多重变异效应分析功能数据模块;将数据提交到ClinGen Linked Data Hub,通过规范等位基因标识符(CAid)实现数据关联;同时与Shariant平台开展试点整合。结果显示,多重变异效应分析数据可在这些平台中与临床变异数据、蛋白结构数据、保守性数据等整合展示,例如在G2P Portal中,TP53的多重变异效应分析数据与ClinVar中的致病突变区域、蛋白DNA结合结构域高度重叠;在UCSC Browser中可直观对比不同药物处理下TP53变异的功能响应差异;在DECIPHER平台中,BRCA1变异的多重变异效应分析数据可辅助临床变异的致病性评估。
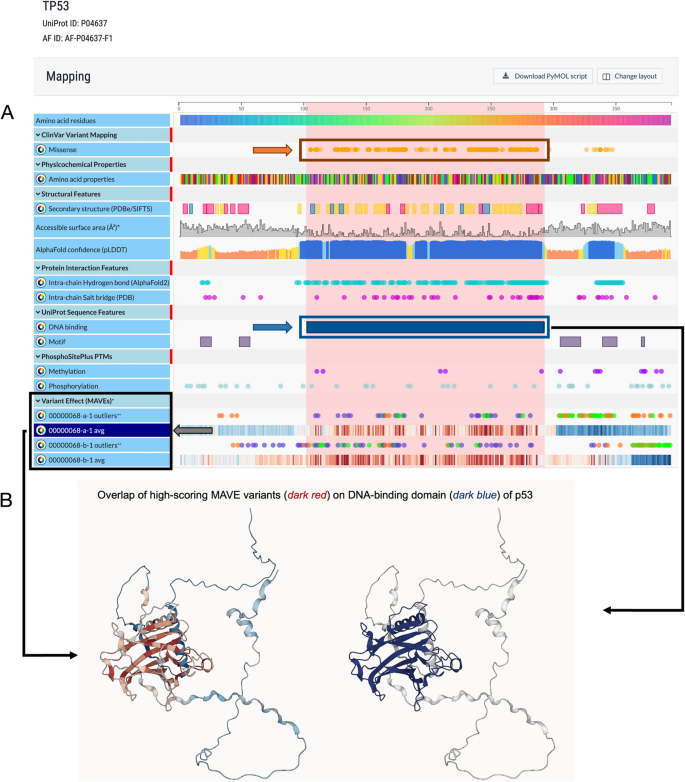
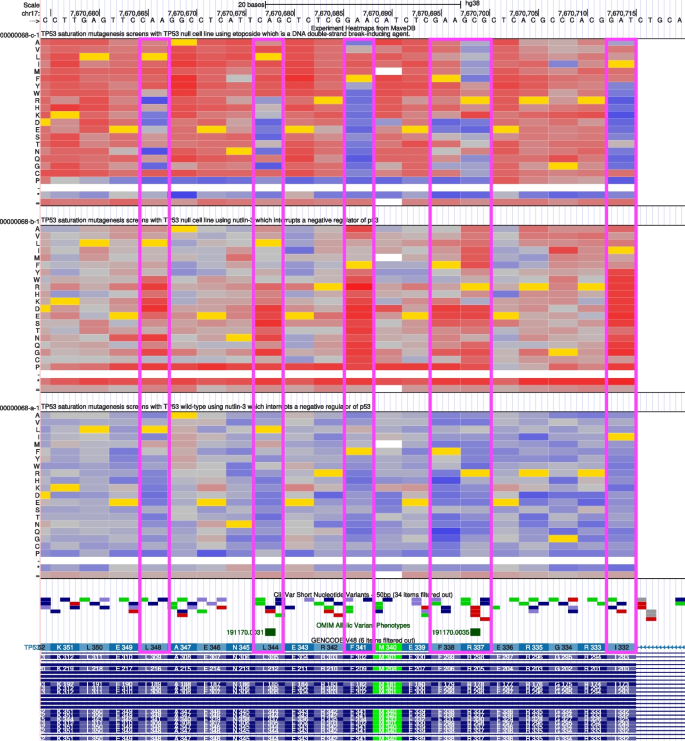
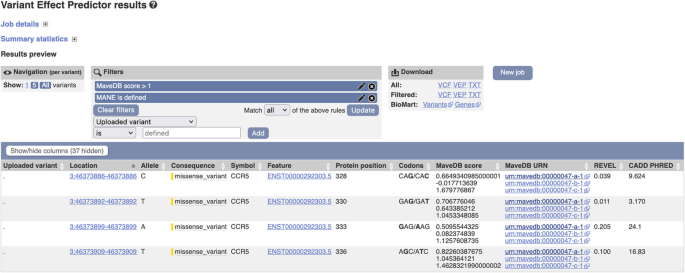
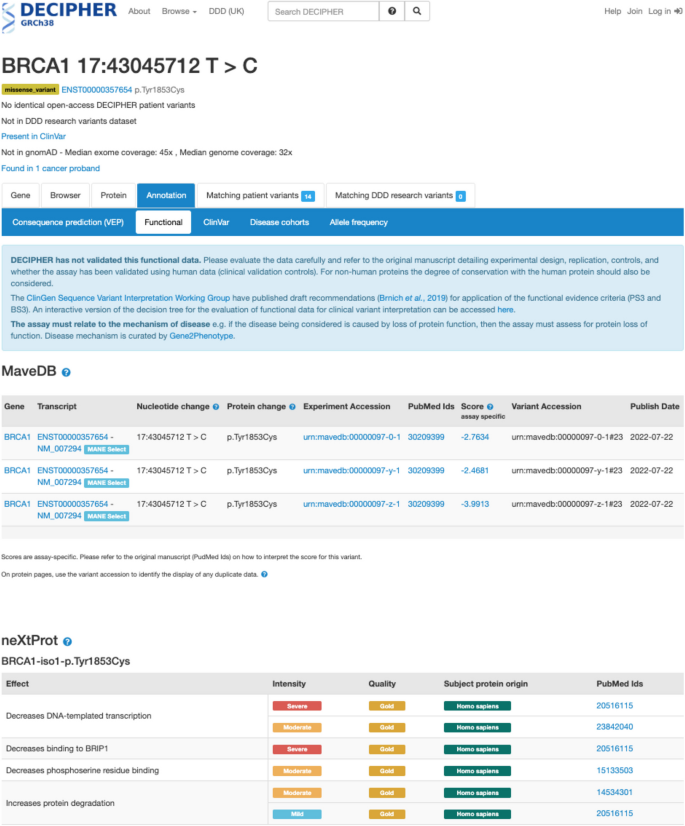
文献未提及具体实验产品,领域常规使用Python、NextFlow等工作流工具进行数据整合与可视化开发。
4. Biomarker研究及发现成果解析
本文的核心成果是建立了多重变异效应分析功能变异数据与人类参考基因组的标准化映射资源,这些功能变异得分可作为辅助临床变异解读的功能Biomarker,其筛选与验证逻辑遵循“MaveDB人类数据集筛选→序列比对验证→变异标准化映射→多平台临床应用验证”的完整链条。
本研究中的Biomarker为多重变异效应分析技术产生的变异功能得分,数据来源于MaveDB中已发表的人类基因功能实验,涵盖临床相关基因如TP53、BRCA1等。验证方法包括通过BLAT序列比对确保多重变异效应分析变异与参考基因组变异的位置一致性,通过VRS标准规范化变异表示以提升数据互操作性。特异性数据显示,一致的变异映射对占总变异数的68.44%,表明大部分多重变异效应分析变异可精准对应到人类参考基因组;敏感性方面,1064个人类数据集中有1057个成功完成映射,映射成功率达99.0%(文献未明确提供统计学显著性数据,基于映射成功率显示方法稳定性较高)。
核心成果方面,本研究建立了目前规模最大的多重变异效应分析数据与人类参考基因组的映射资源,包含超过900万个变异的标准化映射信息,并成功整合到6个主流人类基因组学平台。该资源的功能关联在于,为临床变异解读提供了可及的大规模功能证据,例如可将多重变异效应分析功能得分整合到ACMG/AMP变异解读框架中,辅助意义未明变异的致病性分类;创新性在于首次实现了多重变异效应分析数据的规模化标准化映射与多平台整合,在保留实验数据溯源的同时解决了多重变异效应分析数据与临床基因组学工具的互操作性难题。此外,研究人员开发了可重复的映射工作流,以Python包形式发布,为未来MaveDB新增数据集的自动映射提供了工具支持。