1. 领域背景与文献引入
文献英文标题:Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis;发表期刊:Genome Biology;影响因子:13.214(2017年);研究领域:自身免疫病(强直性脊柱炎)与肠道微生物组
强直性脊柱炎是一种慢性炎症性自身免疫病,以中轴骨骼、外周关节及韧带附着点炎症为核心特征,中国汉族人群患病率为0.2%-0.54%,主要影响青壮年男性的躯体功能与生活质量,给患者和社会带来沉重负担。由于疾病进展隐匿,症状出现到确诊的延迟可达8-10年,当前最有效的肿瘤坏死因子(TNF)抑制剂仅对部分患者有效,无法完全阻止骨侵蚀或韧带骨赘形成,因此深入解析发病机制、开发早期诊断工具是领域核心需求。
近年来,肠道微生物组与自身免疫病的关联成为研究热点,领域共识:肠道微生物组可通过调控先天与适应性免疫系统参与自身免疫病的发生发展。已有研究证实,炎症性肠病、类风湿关节炎等疾病均存在显著的肠道菌群紊乱,但强直性脊柱炎的肠道微生物组研究多局限于16S核糖体RNA测序,分辨率较低,仅能揭示属水平的菌群变化,无法深入到基因与功能层面,且缺乏大样本中国人群的定量研究数据,肠道菌群如何参与强直性脊柱炎发病的具体机制仍不明确。针对这一研究空白,本研究采用定量宏基因组测序技术,系统分析中国人群强直性脊柱炎患者的肠道微生物组特征,筛选特异性生物标志物并构建诊断模型,为疾病的早期诊断与发病机制解析提供新依据。
2. 文献综述解析
作者对领域现有研究的评述逻辑按“疾病临床与遗传背景→肠道微生物组与自身免疫病关联→强直性脊柱炎微生物研究现状→宏基因组技术应用潜力”的维度展开,结构化梳理了三类核心信息。
在强直性脊柱炎基础研究方面,现有研究已明确疾病与HLA-B27基因强相关,且与炎症性肠病存在遗传风险因子重叠,提示两者可能共享相似的发病机制;但现有研究多聚焦于遗传因素,对环境因素尤其是肠道微生物组的作用解析不足。在肠道微生物组与自身免疫病的关联研究中,已有研究证实炎症性肠病、类风湿关节炎等疾病存在显著的肠道菌群紊乱,且宏基因组技术已成功应用于代谢综合征、肝硬化等疾病的微生物组分析,能实现基因与功能层面的深度解析,但强直性脊柱炎领域尚未开展此类定量宏基因组研究。在强直性脊柱炎微生物研究领域,现有16S测序研究发现患者肠道菌群存在普雷沃氏菌科富集、瘤胃球菌科减少等变化,但研究样本量较小,且仅能揭示菌群组成的表层变化,无法明确功能层面的差异及具体发病机制,也缺乏中国人群的针对性数据。
通过对比现有研究的局限,本研究的创新价值凸显:首次采用定量宏基因组测序技术分析大样本中国强直性脊柱炎人群的肠道微生物组,突破了16S测序的分辨率限制,能深入解析基因、功能模块及未培养菌群的变化;同时构建了基于肠道微生物组的诊断模型,为疾病的早期非侵入性诊断提供了新工具,填补了强直性脊柱炎定量宏基因组研究的空白。
3. 研究思路总结与详细解析
本研究以“肠道微生物组紊乱参与强直性脊柱炎发病”为核心科学问题,以“明确中国人群AS肠道菌群特征→筛选特异性生物标志物→构建并验证诊断模型”为研究目标,采用“队列构建→宏基因组测序→多维度组学分析→标志物验证→诊断模型构建”的闭环技术路线,系统解析了AS患者的肠道微生物组变化及潜在功能机制。
3.1 研究队列构建与样本收集
实验目的是建立基线特征匹配、混杂因素控制严格的强直性脊柱炎患者与健康对照队列,确保组间可比性。方法细节:纳入211名中国个体,分为发现队列(73名强直性脊柱炎患者、83名健康对照)和验证队列(24名强直性脊柱炎患者、31名健康对照),患者符合修订的纽约强直性脊柱炎诊断标准,排除合并胃肠道疾病、1个月内使用抗生素的个体;收集所有受试者的粪便样本,采用酚/三氯甲烷法提取微生物DNA。结果解读:队列基线特征分析显示,患者与对照的年龄、体重指数(BMI)无显著差异,排除了年龄、体重对肠道菌群的混杂影响,确保后续菌群差异分析的可靠性。产品关联:文献未提及具体实验产品,领域常规使用粪便DNA提取试剂盒、NanoDrop核酸定量仪、Illumina测序平台等。
3.2 整合基因目录构建与宏基因组测序
实验目的是构建覆盖强直性脊柱炎肠道菌群的整合基因目录,为后续定量分析提供全面的参考数据库。方法细节:对粪便样本进行深度鸟枪法测序,组装强直性脊柱炎患者与对照的肠道微生物基因,将其与已有的人类肠道整合基因目录(IGC)、肝硬化肠道基因目录整合,构建更新版的整合基因目录(IGC2)。结果解读:IGC2包含了约17%的新增基因,能更全面地覆盖中国人群肠道微生物组的基因信息,显著提升了宏基因组数据的比对效率,为后续的菌群组成与功能分析奠定了基础。
3.3 肠道菌群系统发育差异分析
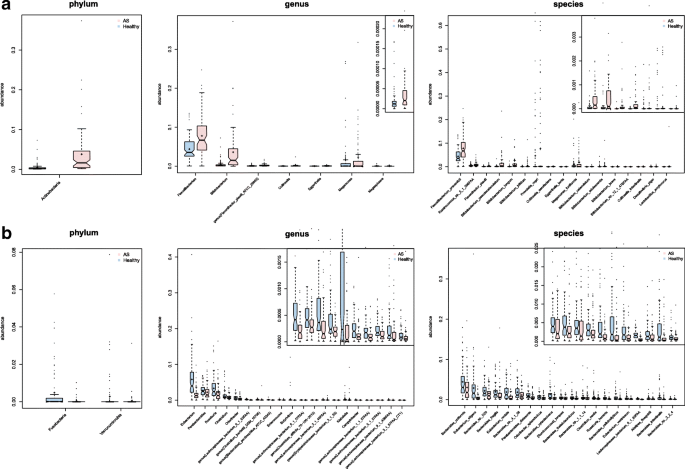
实验目的是比较强直性脊柱炎患者与健康对照在门、属、种水平的菌群组成差异,明确疾病相关的菌群特征。方法细节:将测序reads比对到NCBI与人类微生物组计划(HMP)的参考基因组数据库,采用Wilcoxon秩和检验筛选差异丰度的分类群。结果解读:强直性脊柱炎患者肠道菌群的放线菌门丰度显著升高(n=156,P=1.50e-15),梭杆菌门与疣微菌门丰度显著降低;属水平上,双歧杆菌属、普雷沃氏菌属等富集,拟杆菌属减少;种水平上,Prevotella melaninogenica、Prevotella copri等在患者中显著富集,10种拟杆菌属物种丰度降低。这些结果表明AS患者存在显著的肠道菌群紊乱,且紊乱程度比炎症性肠病更为突出。
3.4 肠道菌群功能与代谢通路分析
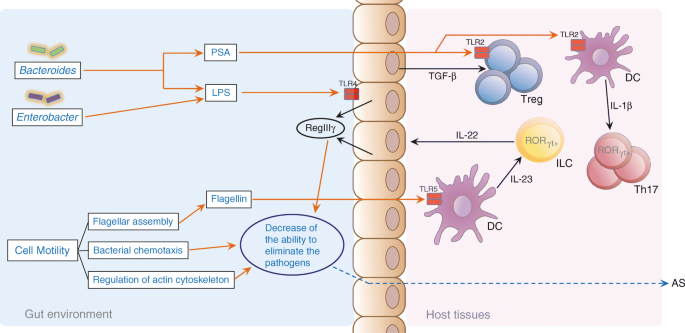
实验目的是解析强直性脊柱炎患者肠道菌群的功能变化,探索菌群参与疾病发病的潜在机制。方法细节:采用京都基因与基因组百科全书(KEGG)对差异基因进行功能注释,筛选差异丰度的KEGG直系同源组(KO)与功能模块。结果解读:AS患者肠道菌群中与膜转运、细胞运动、辅因子和维生素代谢相关的功能模块显著富集,而糖胺聚糖代谢、次级代谢物生物合成、共生相关模块在健康对照中富集;此外,患者肠道菌群的蛋白酶体功能模块显著富集,且所有差异蛋白酶体基因均属于细菌蛋白酶体,这与放线菌门的富集特征一致,提示细菌蛋白酶体可能通过调控NF-κB通路参与炎症反应。
3.5 宏基因组物种与共丰度基因群分析
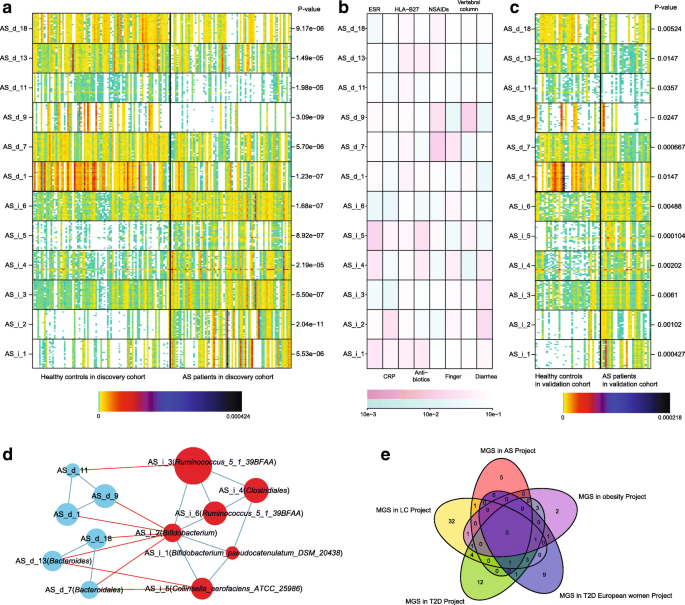
实验目的是筛选稳定的、具有个体间一致性的菌群标志物,减少个体差异对结果的影响。方法细节:将差异丰度基因按丰度相关性聚类为宏基因组物种(MGS),采用共丰度分析构建共丰度基因群(CAG),并在验证队列中验证这些特征的稳定性。结果解读:发现队列中筛选出6个AS富集的MGS和23个健康对照富集的MGS,其中12个MGS在验证队列中仍显著;同时鉴定出199个大CAG和755个小CAG,其中约31.7%的大CAG和77.2%的小CAG为未知分类群,提示AS患者肠道中存在大量未被报道的潜在微生物物种。
3.6 诊断模型构建与验证
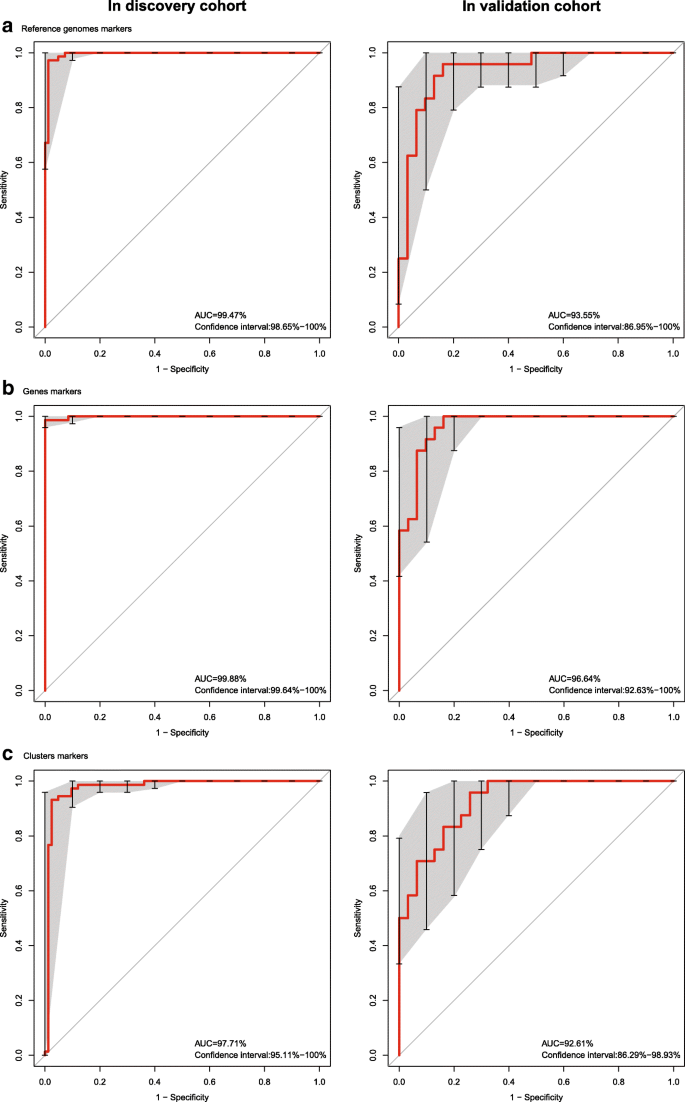
实验目的是基于筛选出的肠道微生物组标志物,构建强直性脊柱炎的非侵入性诊断模型并验证其性能。方法细节:采用支持向量机(SVM)分别基于参考基因组标志物、基因标志物、聚类标志物构建分类器,通过受试者工作特征(ROC)曲线评估模型的诊断性能。结果解读:基因标志物的诊断性能最优,在验证队列中的曲线下面积(AUC)为96.64%(n=55);参考基因组标志物的AUC为93.55%,聚类标志物的AUC为92.61%,三类模型均显示出良好的诊断效能,表明肠道微生物组可作为强直性脊柱炎的有效诊断生物标志物。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究共筛选出三类强直性脊柱炎相关生物标志物,包括系统发育水平的分类群标志物(门、属、种水平的差异菌群)、基因水平的差异丰度基因标志物、宏基因组物种(MGS)与共丰度基因群(CAG)标志物。筛选逻辑为:首先在发现队列中通过Wilcoxon秩和检验筛选具有统计学差异的菌群特征,再在独立验证队列中验证其稳定性,确保标志物的可靠性与重复性。
研究过程详述
所有标志物均来源于211名中国个体的粪便样本,验证方法包括宏基因组测序定量、Wilcoxon秩和检验的统计学验证、支持向量机分类器的诊断性能验证。特异性与敏感性方面,基因标志物在验证队列中的AUC为96.64%(n=55),显示出极高的诊断特异性;参考基因组标志物的AUC为93.55%,聚类标志物的AUC为92.61%,三类标志物均能有效区分强直性脊柱炎患者与健康对照。
核心成果提炼
本研究发现的生物标志物不仅具有临床诊断价值,还揭示了潜在的发病机制:AS患者中富集的双歧杆菌属部分物种可通过分子模拟激活Th2型免疫反应,普雷沃氏菌属物种可能通过刺激免疫反应靶向关节组织;蛋白酶体功能模块的富集提示细菌蛋白酶体可能通过调控IκB-α的泛素化激活NF-κB通路,促进炎症因子积累。此外,本研究首次在大样本中国人群中明确了强直性脊柱炎的肠道菌群紊乱特征,构建的诊断模型为疾病的早期非侵入性诊断提供了新工具,同时为开发靶向肠道微生物组的强直性脊柱炎治疗方法提供了新靶点。