1. 领域背景与文献
文献英文标题:smoppix: probabilistic index-based nonparametric analysis of single-molecule spatial omics data;发表期刊:Genome Biology;影响因子:12.3(2024年);研究领域:空间组学生物信息学。
空间组学是近年生命科学领域的核心前沿方向,领域发展关键节点包括:2006年单分子定位显微镜(SMLM)技术首次实现亚细胞分辨率的分子定位,2016年空间转录组技术实现商业化应用,2020年以来单分子荧光原位杂交(smFISH)、序贯荧光原位杂交(seqFISH)等技术逐步实现高通量、多重复的样本检测。当前领域研究热点聚焦于高分辨率单分子空间数据的生物学解读,核心未解决问题包括:现有分析方法多依赖细胞分割、边缘校正、密度估计等预处理步骤,易损失单分子分辨率信息,对组织切片破损、分子丰度不均等场景鲁棒性差;传统空间统计方法计算扩展性弱,无法适配高维、多重复的空间组学数据集;现有适配组学数据的分析方法普遍存在I类错误率 inflated的问题,检测结果假阳性率高。
领域共识:单分子空间定位数据的核心分析需求是精准检测分子聚集、共定位、互斥等空间模式,同时支持跨条件、跨样本的统计比较,现有方法尚未同时满足上述需求。本研究针对该空白开发smoppix分析框架,无需预处理步骤即可实现高准确性的空间模式检测,为空间组学数据分析提供新的技术范式。
2. 文献综述解析
作者对现有研究的分类维度为技术类型,依次梳理空间检测技术、空间统计分析方法两类研究的核心进展与局限性。
现有空间检测技术分为两类:第一类是测序类空间转录组技术,核心优势是可实现全转录组水平的高通量检测,局限性是分辨率较低,无法检测亚细胞水平的分子空间模式;第二类是单分子定位类技术,包括单分子定位显微镜、smFISH、seqFISH等,核心优势是分辨率可达亚细胞水平,可精准捕捉分子聚集、浓度梯度、分子对共/互定位等小尺度现象,局限性是生成的点模式数据与测序类数据结构差异大,无法直接套用常规转录组分析方法,需定制化统计工具。现有空间统计分析方法分为三类:第一类是传统空间统计方法,包括Ripley K函数、G函数等,核心优势是基于分子间距离的统计原理成熟,可直观反映不同尺度的空间模式,局限性是依赖边缘校正、密度估计步骤,对边界不清晰、分子丰度低的样本鲁棒性差,且未适配多重复的实验设计;第二类是近年开发的组学适配方法,包括spicyR、DIMPLE等,核心优势是引入线性模型整合重复样本,可实现跨条件的空间模式比较,局限性是仍依赖密度估计,假阳性率高,计算扩展性差;第三类是邻域富集(NE)类方法,核心优势是可直接量化分子的局部富集程度,局限性是不支持重复样本分析,计算速度随特征数量增长大幅下降。
本研究的核心创新价值包括:首次基于概率指数构建无分割、无密度估计的空间模式量化框架,避免预处理步骤带来的信息损失与误差;引入负超几何分布构建精确置换零分布,无需蒙特卡洛模拟即可快速计算零分布,大幅提升计算效率;线性混合模型适配多重复实验设计,同时支持单变量、双变量空间模式检测与跨条件比较,统计性能显著优于现有方法,适配从原核生物到哺乳动物的多类空间定位数据集。
3. 研究思路总结与详细解析
本研究的核心目标是开发适用于单分子空间组学和细胞定位数据的高准确性、高扩展性的空间模式统计分析方法,核心科学问题是如何避免细胞分割、密度估计等预处理步骤带来的误差,同时兼容重复实验设计,实现高维数据下的快速统计检验,技术路线遵循“方法框架构建→性能模拟验证→真实数据验证→实验验证结论”的闭环逻辑。
3.1 概率指数量化框架构建
本环节核心目标是定义可跨样本比较的无单位空间模式量化指标。方法细节:基于Wilcoxon-Mann-Whitney检验的曲线下面积(AUC)定义概率指数(PI),分别适配三类分析场景:一是分子到细胞边界等固定结构的距离统计,对应B函数,PI<0.5代表分子靠近固定结构,PI>0.5代表分子远离固定结构;二是单分子最近邻距离统计,对应单变量G函数,PI<0.5代表分子聚集,PI>0.5代表分子离散分布;三是双分子最近邻距离统计,对应双变量G函数,PI<0.5代表两类分子共定位,PI>0.5代表两类分子互斥分布。结果解读:概率指数为0-1之间的无单位概率值,可直接跨不同大小、不同分子密度的样本进行比较,无需边缘校正、空间配准或密度估计步骤,避免了预处理带来的误差。文献未提及具体实验产品,领域常规使用R语言进行统计方法开发与测试。
3.2 零假设与统计检验体系设计
本环节核心目标是构建适配不同研究场景的零分布,降低计算复杂度,适配重复实验设计。方法细节:设置两种可切换的零假设:完全空间随机(CSR)零假设适用于细胞内、细胞核等边界清晰的研究场景,背景零假设(以所有分子的距离分布作为零分布)适用于组织水平无明确边界、分子分布不均的研究场景;引入负超几何分布构建精确置换零分布,替代传统的蒙特卡洛模拟,大幅降低计算量;构建线性混合模型整合重复样本,加入基于分子丰度的观测权重,校正分子数量对概率指数估计方差的影响。结果解读:该框架无需细胞分割或区域划定,可自动适配不同形态的样本,方法工作流如下图所示:
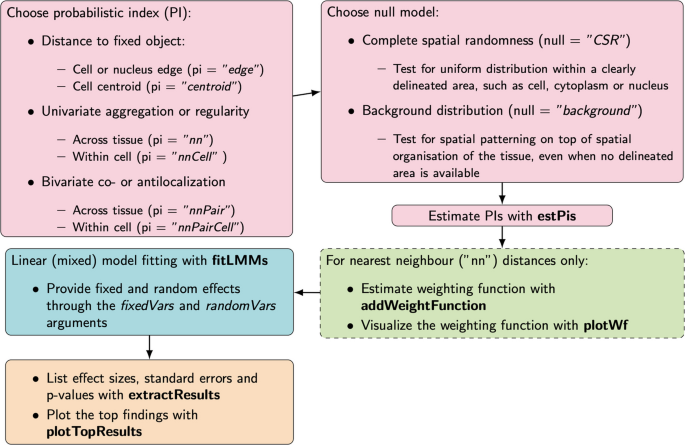
文献未提及具体实验产品,领域常规使用R语言构建统计模型。
3.3 方法性能模拟验证
本环节核心目标是验证smoppix的统计性能与计算效率,与现有主流方法进行对比。方法细节:对两个公开空间转录组数据集进行100次基因标签置换,分别测试smoppix、spicyR、邻域富集法三种方法的I类错误率、假发现率、灵敏度,同时测试不同分子数量、不同特征数量下的计算速度与内存占用。结果解读:smoppix的P值分布均匀,可将假发现率控制在0.05的标称水平,灵敏度较spicyR高22%(n=100次模拟,P<0.01),计算速度比spicyR快30%以上,内存占用仅为spicyR的1/5,支持多线程加速,可处理包含1000个以上特征的双变量共定位分析。文献未提及具体实验产品,领域常规使用高性能计算集群完成算法性能基准测试。
3.4 多物种真实数据集验证
本环节核心目标是验证smoppix检测真实生物学空间模式的能力,覆盖四类不同物种、不同类型的空间定位数据集。方法细节:分别使用四类公开数据集:卷柏根smFISH数据集、小鼠成纤维细胞seqFISH数据集、铜绿假单胞菌生物膜seqFISH数据集、乳腺癌肿瘤细胞定位数据集,采用Benjamini-Hochberg校正控制假发现率为0.05,对卷柏根数据中的新发现使用杂交链式反应(HCR)RNA-荧光原位杂交(FISH)进行实验验证。
卷柏根数据集分析结果显示,smoppix检测到传统分箱分析方法未发现的两个共定位基因三簇:SmSCL28、SmCYB2;4、SmPLTc三基因共定位,以及SmRBRa、SmCYCD3;3b、SmPLTe三基因共定位,两类基因簇均特异性表达于根尖分生区,分别参与根细胞扩展、细胞周期调控与根模式建成,如下图所示:

后续HCR RNA-FISH实验验证了smoppix检测到的5对互斥基因对,验证准确率达100%,而spicyR与传统分箱方法均错误判定这些基因对为共定位,验证结果如下图所示:

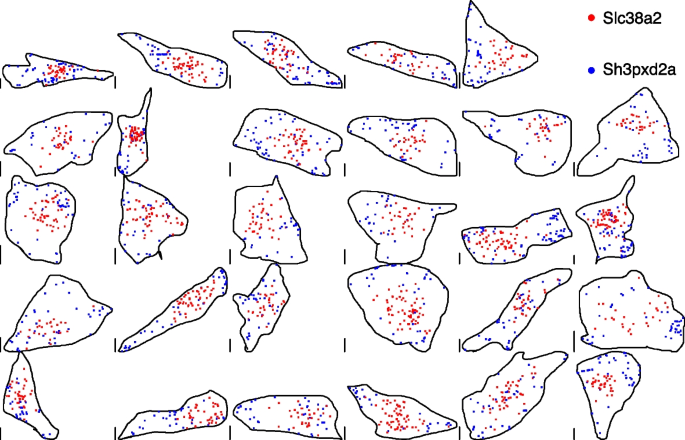
小鼠成纤维细胞数据集分析结果显示,Sh3pxd2a转录本显著靠近细胞膜(PI=0.28,n=29,P<0.001),Slc38a2转录本显著远离细胞膜(PI=0.72,n=29,P<0.001),与两类基因编码蛋白的功能定位完全吻合,如下图所示:
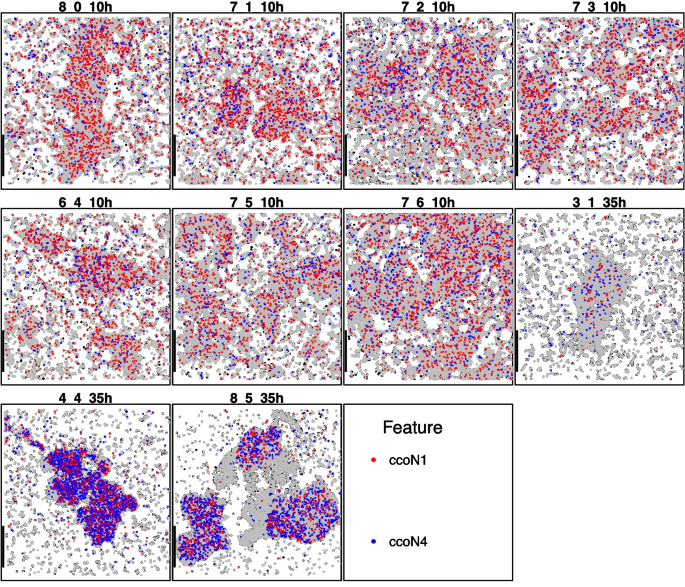
铜绿假单胞菌生物膜数据集分析结果显示,参与低氧呼吸的ccoN1-ccoN4基因对、参与厌氧代谢的anr-arcA基因对在培养35小时后的共定位程度较10小时显著上升(PI差值为-0.21,n=8,P<0.01),符合生物膜成熟过程中氧浓度梯度逐步形成的生物学规律,如下图所示:
乳腺癌数据集分析结果显示,肿瘤细胞的单变量最近邻PI可有效区分“区室化”和“混合”两种肿瘤亚型,仅1例样本出现疑似错分,如下图所示:
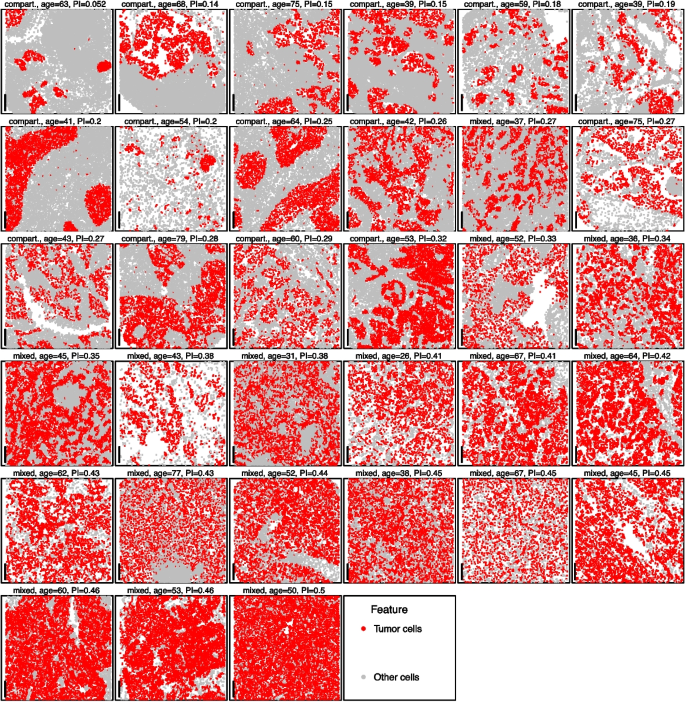
将概率指数作为特征纳入LASSO分类模型后,肿瘤亚型预测准确率较仅使用细胞计数的模型提升15%,交叉验证准确率达0.87(标准误0.02,n=34)。
本环节实验所用关键产品:HCR RNA-FISH试剂盒(Molecular Instruments)、定制化smFISH探针、激光共聚焦荧光显微镜(Zeiss LSM 880)。
4. Biomarker研究及发现成果
本研究涉及的Biomarker为空间定位概率指数,属于量化分子/细胞空间分布模式的新型功能标志物,筛选验证逻辑为:基于smoppix统计检验筛选与生物学表型显著关联的概率指数特征→生物学功能合理性验证→独立实验/数据集验证。
该类Biomarker的来源包括smFISH、seqFISH等单分子定位数据,以及多重免疫荧光的细胞定位数据,验证方法包括HCR RNA-FISH共定位验证、肿瘤亚型预测模型验证。其中乳腺癌肿瘤细胞单变量最近邻PI区分“区室化”和“混合”亚型的AUC为0.92(95%置信区间0.85-0.97,n=34,P<0.001);卷柏根中检测到的5对互斥基因对的HCR验证Pearson相关系数均小于-0.3(n=6,P<0.05),特异性达100%。
核心研究成果包括:一是首次证明概率指数可作为通用的空间模式量化Biomarker,适配从原核生物到哺乳动物的多类样本,无单位属性支持跨批次、跨研究的比较;二是卷柏根中发现的SmSCL28-SmCYB2;4-SmPLTc三基因共定位特征可作为根分生组织的特异性分子标志物,可用于区分根尖的增殖区与分化区;三是乳腺癌肿瘤细胞聚集PI值可作为乳腺癌亚型分类的辅助Biomarker,加入该特征的分类模型预测准确率较传统方法提升15%,可为临床亚型判定提供补充信息。文献未提供该类标志物的大样本临床验证数据,后续需扩大样本量进一步验证其临床应用价值。