1. 领域背景与文献引入
文献英文标题:KcatNet: a geometric deep learning framework for genome-scale prediction of enzyme turnover numbers;发表期刊:Genome Biology;影响因子:17.906;研究领域:计算酶学、代谢组学与合成生物学交叉领域。
酶周转数(Kcat)是表征酶催化效率的核心动力学参数,在基因组尺度代谢模型(GEMs)构建、酶进化机制解析、合成生物学酶工程设计中具有不可替代的作用。领域共识:实验测量Kcat存在通量低、成本高、依赖酶纯化等瓶颈,目前仅约12%的大肠杆菌酶反应有实验Kcat数据。现有计算工具虽实现了高通量预测,但普遍存在泛化性不足、对高Kcat值预测偏差大、缺乏结构层面可解释性的问题,无法满足代谢建模和酶工程的精准需求。针对这一研究空白,本研究开发了KcatNet,一种整合预训练蛋白语言模型序列特征与几何深度学习结构特征的预测工具,旨在实现全基因组尺度的准确Kcat预测,并提供残基水平的催化机制解释。
2. 文献综述解析
作者按机器学习框架类型对现有Kcat预测工具进行分类,涵盖梯度提升、额外树、注意力机制等模型,系统梳理了各工具的技术路线与性能局限。
现有Kcat预测工具如DLkcat、TurNuP、UniKP等的核心优势是突破了实验测量的通量限制,实现了跨物种的酶-底物对Kcat高通量预测,为代谢建模提供了大规模参数支持。但这些工具存在明显局限性:一是对高Kcat值的预测准确性差,由于实验数据中高Kcat样本占比低,模型易出现偏差;二是泛化性不足,对与训练集序列相似度低的酶预测性能显著下降;三是缺乏可解释性,无法从结构层面解析Kcat差异的分子机制。
通过对比现有工具的不足,KcatNet的创新价值凸显:首次将预训练蛋白语言模型的序列特征与几何深度学习的结构特征整合,既提升了预测准确性尤其是高Kcat值,又实现了残基水平的可解释性,为酶催化机制解析和定向进化提供了新的技术范式。
3. 研究思路总结与详细解析
本研究的核心目标是构建兼具高准确性、强泛化性和可解释性的全基因组尺度Kcat预测工具,核心科学问题是如何整合酶序列与底物结构特征,实现精准的Kcat预测并解析催化机制,技术路线遵循“数据集构建→模型设计→性能验证→可解释性分析→应用拓展”的闭环逻辑。
3.1 数据集构建与预处理
实验目的是构建高质量、无偏的Kcat训练与测试数据集,为模型训练提供可靠基础。方法细节上,研究整合了BRENDA、SABIO-RK等数据库的野生型和突变体Kcat数据,对超过半数的数据点进行手动验证以确保数值与原始文献一致,排除噪声数据后将所有Kcat值转换为对数尺度,最终将数据集按8:2比例划分为训练集和测试集。结果解读显示,最终得到11757个有效条目,包含11288个独特反应和7441个独特酶,数据集覆盖了跨物种、跨酶家族的多样本类型,为模型泛化性验证提供了基础。产品关联:文献未提及具体实验产品,领域常规使用数据库检索工具、Python数据清洗脚本等完成数据集构建。
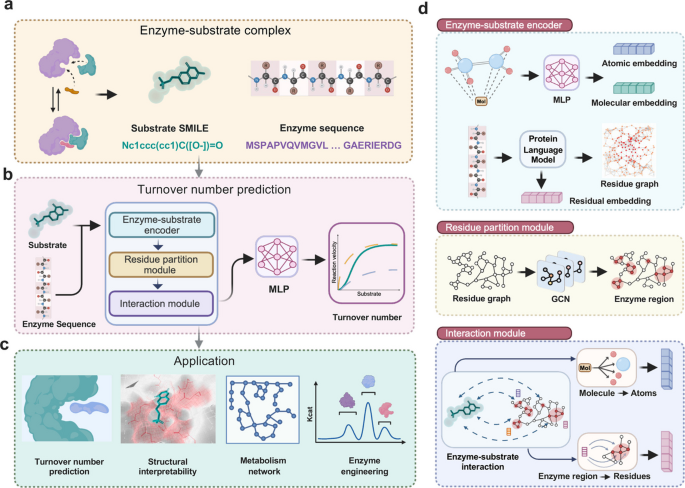
3.2 KcatNet模型架构设计
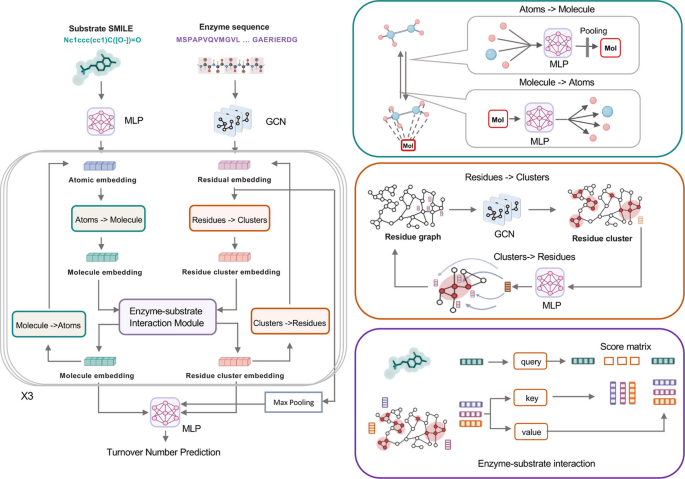
实验目的是构建能够同时整合酶序列信息与底物结构信息的深度学习模型,实现准确且可解释的Kcat预测。方法细节上,模型包含三个核心模块:酶-底物编码器模块,采用ProtT5和ESM2两种预训练蛋白语言模型生成酶残基水平的特征嵌入,同时通过SMILES transformer实现底物的原子级和分子级特征编码;基于图的残基分区模块,利用图卷积神经网络(GCN)将酶残基按空间邻近性分组,捕捉酶的局部结构特征;催化复合物周转数预测器模块,通过注意力机制建模酶残基分区与底物的相互作用模式,结合消息传递和最大池化操作输出最终Kcat预测值。结果解读显示,该架构实现了序列特征与结构特征的有机融合,能够从分子层面捕捉酶-底物相互作用对催化效率的影响。产品关联:文献未提及具体实验产品,领域常规使用PyTorch、TensorFlow等深度学习框架完成模型开发。
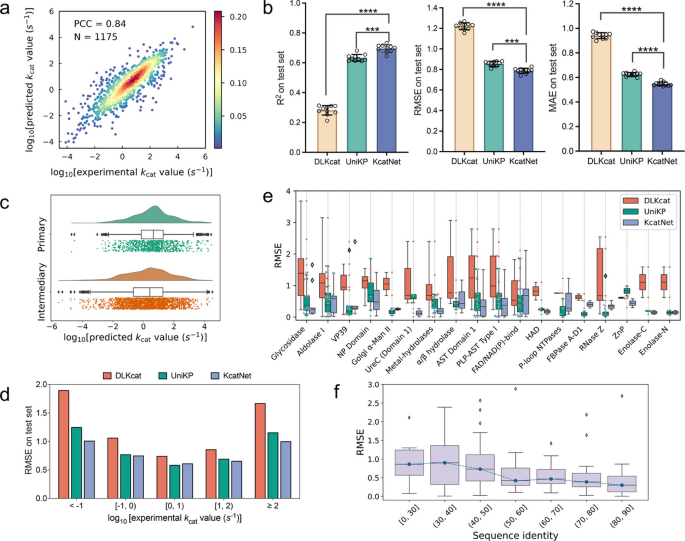
3.3 模型性能评估
实验目的是全面验证KcatNet的预测准确性、泛化性以及对不同Kcat范围的预测能力。方法细节上,在测试集上计算皮尔逊相关系数(PCC)、决定系数(R²)、均方根误差(RMSE)等指标,并与DLkcat、UniKP等主流工具进行对比;同时分层评估模型在不同Kcat范围、不同蛋白家族、不同物种以及突变体酶中的预测性能。结果解读显示,KcatNet在测试集上的PCC达0.84,RMSE为0.78,显著优于现有工具(与DLkcat相比,RMSE降低具有统计学显著性,P=4.35×10⁻¹⁰);尤其在高Kcat值预测上表现突出,RMSE稳定在0.61-1.01之间,预测值与实验值相差不超过一个数量级;对与训练集序列相似度低的酶,模型仍能保持较高预测准确性,泛化性显著提升。产品关联:文献未提及具体实验产品,领域常规使用Scikit-learn、Matplotlib等工具完成性能评估与可视化。
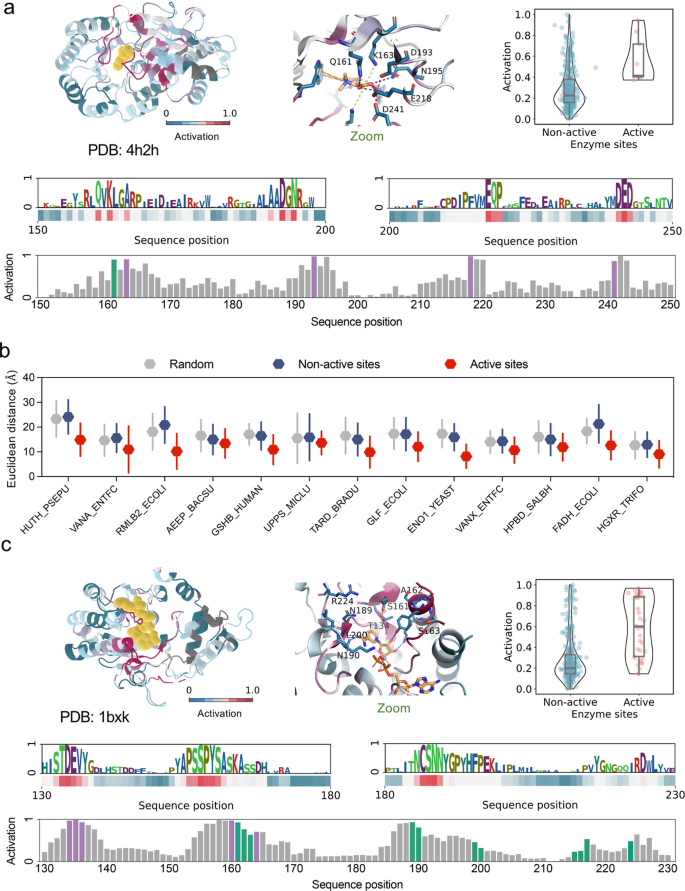
3.4 模型可解释性分析
实验目的是解析KcatNet预测的分子基础,识别与催化效率相关的关键残基。方法细节上,通过计算模型的残基注意力权重(激活得分),将高得分残基映射到酶的三维结构,并与M-CSA数据库中的已知催化残基进行空间距离统计;同时结合碱性磷酸酶的高通量突变数据,验证模型对远程变构残基的识别能力。结果解读显示,KcatNet识别的高得分残基集中在催化口袋附近,与已知催化残基的空间距离显著小于低得分残基;此外,模型还能识别远程变构残基,这些残基的突变会显著影响催化效率,证明模型能够捕捉酶催化的全尺度结构机制。产品关联:文献未提及具体实验产品,领域常规使用PyMOL、UCSF Chimera等结构可视化工具完成残基映射分析。
3.5 代谢建模应用验证
实验目的是验证KcatNet预测值对基因组尺度代谢模型性能的提升作用。方法细节上,将KcatNet预测的Kcat值整合到四种酵母的酶约束代谢模型(ecGEMs)中,对比原始模型、DLkcat参数化模型与KcatNet参数化模型的生长率预测准确性,评估指标为均方根误差(RMSE)。结果解读显示,KcatNet参数化的模型在16/22的环境-物种组合中降低了生长率预测的RMSE,显著提升了代谢模型对蛋白分配和细胞生长的预测能力。产品关联:文献未提及具体实验产品,领域常规使用COBRApy等代谢建模工具完成模型构建与模拟。
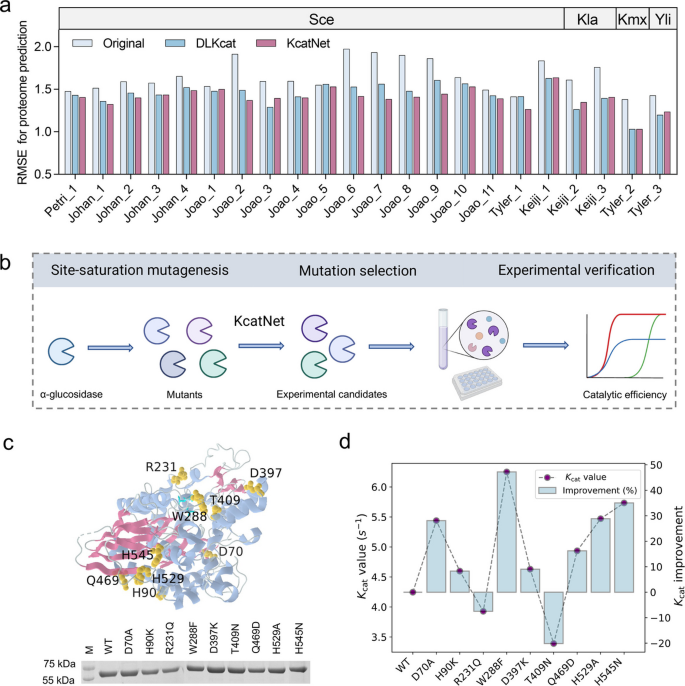
3.6 酶工程应用验证
实验目的是验证KcatNet指导酶定向进化的实际应用价值。方法细节上,用KcatNet对α-葡萄糖苷酶的10545个单点突变体进行虚拟筛选,选择9个预测Kcat值提升的突变体进行实验验证;通过质粒构建、蛋白表达纯化、酶动力学测定等步骤,检测突变体的Kcat值。结果解读显示,7/9的突变体Kcat值高于野生型酶,证明KcatNet能够有效筛选出具有更高催化效率的酶突变体,为酶定向进化提供了精准靶点。产品关联:实验所用关键产品:Vazyme的2×Phanta Max Mix、GE Healthcare的镍亲和柱、Cytiva的PD-10脱盐柱、Beyotime的Bradford蛋白测定试剂盒。
4. Biomarker研究及发现成果解析
本研究中涉及的Biomarker为与酶催化效率相关的功能残基,包括催化口袋残基和远程变构残基,通过KcatNet的注意力机制筛选并验证,为酶催化机制解析和定向进化提供了精准分子靶点。
Biomarker定位:本研究识别的Biomarker属于酶催化效率相关的功能残基,分为直接参与催化的活性位点残基、参与底物结合的口袋残基以及远程调控催化的变构残基。筛选与验证逻辑为:首先通过KcatNet的注意力权重得分识别对Kcat预测贡献大的残基,然后将这些残基映射到酶的三维结构,与M-CSA数据库的已知催化残基进行空间距离验证,最后通过高通量突变实验验证残基功能的重要性。
研究过程详述:Biomarker来源于酶的全序列残基,验证方法包括结构映射分析和实验功能验证:结构映射分析中,对多个酶的残基注意力得分与已知催化残基的空间距离进行统计,结果显示高得分残基与催化残基的平均距离显著小于低得分残基(文献未提供具体数值);实验功能验证中,在α-葡萄糖苷酶的9个预测突变体中,7个突变体的Kcat值显著高于野生型(文献未提供具体P值和样本量),证明这些残基对催化效率的关键调控作用。
核心成果提炼:该Biomarker的功能关联为直接或间接调控酶的催化效率,突变这些残基会导致Kcat值的显著变化;创新性在于首次通过几何深度学习实现了全基因组尺度的酶催化功能残基高通量识别,突破了传统实验方法的通量限制;在α-葡萄糖苷酶的验证中,7/9的预测突变体获得了更高的Kcat值,证明了Biomarker的实用性,为酶定向进化提供了精准靶点,加速了酶工程的研发进程。