1. 领域背景与文献引入
文献英文标题:2passtools: two-pass alignment using machine-learning-filtered splice junctions increases the accuracy of intron detection in long-read RNA sequencing;发表期刊:Genome Biology;影响因子:13.583(2021年);研究领域:长读长RNA测序中的剪接位点检测与转录组分析。
长读长RNA测序技术(如Oxford Nanopore直接RNA测序(DRS)、PacBio IsoSeq)是解析复杂转录组的核心工具——其能直接测序全长转录本,完整捕捉可变剪接、转录起始/终止位点等信息,解决了短读长RNA测序(Illumina)的“拼接碎片化”问题。然而,长读长的高错误率(尤其是均聚物区域的碱基识别错误)严重影响剪接junction(剪接供体与受体位点的组合区域)的准确识别,导致内含子检测假阳性率高,进而干扰转录本组装与定量的准确性。当前研究热点集中在开发针对长读长的高精度比对算法,但未解决的核心问题是:现有方法要么依赖完整参考注释(无注释时效果差),要么无法有效过滤远距离比对错误,导致内含子检测准确性不足。
本研究的初衷正是针对这一痛点:开发一种不依赖完整参考注释的双次比对方法,结合机器学习过滤假阳性剪接junction,提升长读长RNA测序中的内含子检测准确性。其学术价值在于,为长读长转录组分析提供了更准确的工具(2passtools),不仅能提高已知剪接位点的检测精度,还能在注释不完整的情况下发现novel剪接事件,推动全长转录本的功能研究(如疾病相关的异常可变剪接)。
2. 文献综述解析
文献综述围绕“技术发展-现有方法的优势与局限-未解决问题”展开核心评述:
作者首先对比短读长与长读长RNA测序的技术特征——短读长的“拼接碎片化”促使长读长成为研究可变剪接的关键工具,但长读长的高错误率带来了新挑战:剪接junction识别不准确。随后,作者系统评述了现有解决方案的优缺点:(1)后比对校正方法(如FLAIR)通过参考注释校正剪接junction,但无法校正与参考位点距离较远的错误(如短外显子的比对失败);(2)参考注释引导比对(如minimap2的--junc-bonus参数)能提高已知位点的准确性,但依赖完整注释,非模式生物或注释不完整样本(如疾病组织)中效果有限;(3)metrics过滤方法(如Portcullis)通过junction的支持读长数、motif等过滤假阳性,但未整合序列特征,准确性仍有提升空间。
现有研究的关键结论可总结为:长读长的“全长优势”是其核心价值,但高错误率导致剪接junction识别成为瓶颈;现有方法无法同时解决“无注释依赖”与“高准确性”的问题。
本研究的创新价值在于:(1)提出双次比对+机器学习过滤策略——第一次比对提取junction,机器学习过滤假阳性,第二次比对用过滤后的junction引导,不依赖完整注释也能提高准确性;(2)整合比对metrics与序列特征——将junction的比对距离(JAD)、支持读长数与剪接位点的基因组序列特征结合,训练机器学习模型,比单一metrics或序列模型更全面;(3)开发开源工具2passtools,实现从比对到junction过滤的全流程自动化,提升方法的可重复性。
3. 研究思路总结与详细解析
3.1 整体框架概括
研究目标:开发提高长读长RNA测序内含子检测准确性的双次比对方法;
核心科学问题:如何有效过滤假阳性剪接junction,提升比对准确性;
技术路线:长读长数据获取与模拟→第一次比对提取剪接junction→机器学习过滤假阳性→第二次比对引导→多维度验证(模拟/真实数据、转录组组装)→注释辅助发现novel剪接事件。
3.2 长读长数据获取与模拟
实验目的:获取真实长读长数据,并模拟带已知剪接位点的数据集作为金标准,确保方法性能评估的可靠性。
方法细节:(1)真实数据:获取Arabidopsis Col-0的Nanopore DRS数据(PRJEB32782)、人类GM12878细胞的Nanopore DRS/cDNA数据、小鼠的Nanopore数据(PRJEB27590)及PacBio IsoSeq数据;(2)模拟数据:基于参考转录组(如Arabidopsis的AtRTD2、人类的GRCh38),模拟长读长的长度分布、均聚物错误及随机错误,确保模拟数据的错误特征与真实数据一致。
结果解读:模拟数据的比对错误特征(如短外显子的比对失败、剪接junction假阳性)与真实数据高度一致(图S1),可作为后续验证的金标准。
产品关联:文献未提及具体实验产品,领域常规使用Oxford Nanopore MinION/GridION测序仪、PacBio Sequel II系统进行长读长测序;数据处理常用minimap2比对、StringTie2组装。
3.3 第一次比对与剪接junction提取
实验目的:从长读长数据中提取初始剪接junction,为后续过滤提供基础。
方法细节:使用minimap2进行第一次比对,参数根据数据类型调整:(1)Nanopore DRS数据:-k14 -x splice -L --cs=long;(2)Nanopore cDNA与PacBio数据:-x splice -L --cs=long;(3)设置最大内含子长度(Arabidopsis为10000nt,人类/小鼠为200000nt)以匹配物种特征。从比对结果中提取junction的关键metrics:① JAD(junction alignment distance,剪接junction两侧比对匹配的最短长度);② 支持读长数;③ 内含子 motif(是否为GU/AG等canonical motif);④ primary donor/acceptor(20nt窗口内支持读长数最高的剪接位点)。
结果解读:第一次比对提取到大量剪接junction(如Arabidopsis真实数据提取17521个未注释junction),但仅20%的未注释junction得到Illumina RNAseq支持,说明大部分为假阳性,需进一步过滤。
产品关联:使用minimap2软件进行比对,文献未提及具体商业产品。
3.4 机器学习模型训练与剪接junction过滤
实验目的:训练机器学习模型过滤假阳性剪接junction,得到高质量的引导junction集。
方法细节:(1)第一次过滤:用决策树模型(基于JAD、primary donor/acceptor、内含子 motif)过滤,保留JAD≥4nt、为primary位点且含canonical motif的junction;(2)序列特征模型:提取剪接位点的128nt基因组序列(中心为供体/受体位点),one-hot编码后训练逻辑回归(LR)模型,预测剪接位点的真实性;(3)第二次过滤:用整合JAD、primary位点及LR预测分数的决策树模型,进一步过滤假阳性。
结果解读:整合模型的过滤效果显著优于单一模型——模拟数据中,Arabidopsis的剪接junction检测F1分数从0.732提升至0.954,人类从0.444提升至0.957(图4c);真实数据中,过滤后的junction与Illumina RNAseq的重叠率达57%(未过滤时仅20%),假阳性率显著降低。
产品关联:使用scikit-learn库训练机器学习模型,文献未提及具体商业产品。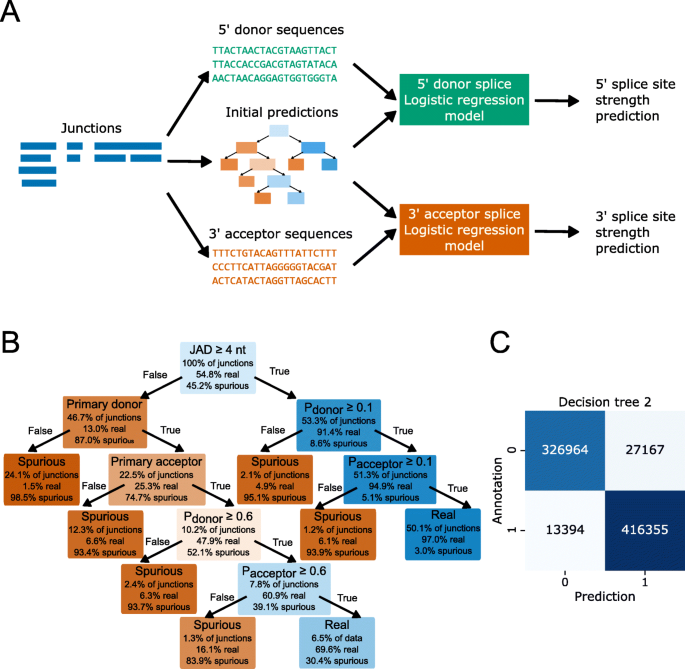
3.5 第二次比对与效果验证
实验目的:用过滤后的剪接junction引导第二次比对,验证方法对内含子检测准确性的提升。
方法细节:(1)第二次比对:使用minimap2,加入--junc-bonus=12参数(引导比对),用过滤后的junction作为引导集;(2)验证指标:① 正确比对率(模拟数据中,比对结果与参考转录本剪接junction一致的比例);② 转录本定量相关性(模拟数据中,预测丰度与真实丰度的Spearman相关系数);③ 转录组组装效果(用StringTie2组装,评估precision(组装转录本与参考一致的比例)与recall(参考转录本被组装到的比例))。
结果解读:(1)正确比对率显著提升:Arabidopsis模拟数据从73.2%提升至89.3%,人类从44.4%提升至65.7%(图5a);(2)转录本定量相关性提高:Arabidopsis的Spearman系数从0.876提升至0.916,人类从0.778提升至0.859(图5b);(3)转录组组装效果提升:Arabidopsis真实数据的precision中位数提高7.1%,人类提高3.5%(图6b-c)。
产品关联:使用StringTie2进行转录组组装,文献未提及具体商业产品。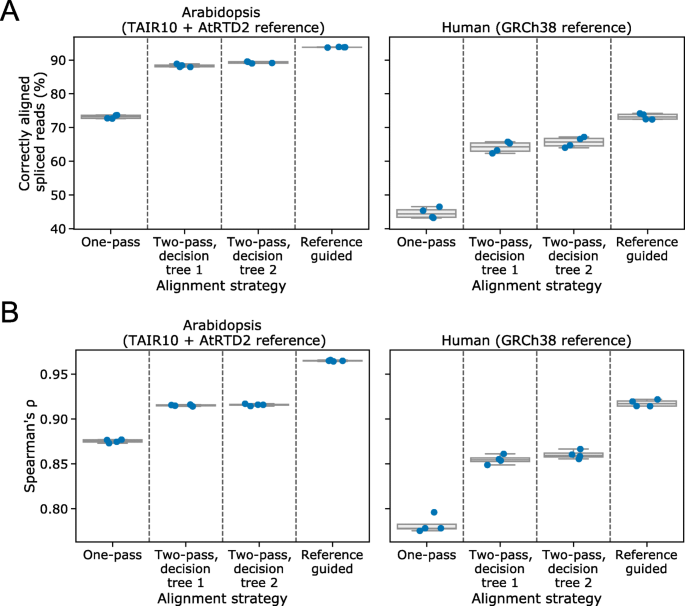
3.6 注释辅助双次比对与novel剪接事件发现
实验目的:在参考注释不完整的情况下,利用现有注释辅助双次比对,发现novel剪接事件。
方法细节:(1)注释辅助模型:用不完整的参考注释(模拟25%~66%的注释缺失)训练随机森林模型,过滤剪接junction;(2)真实样本验证:用Arabidopsis hen2-2突变体(核RNA外切体突变体,积累未注释转录本)的Nanopore DRS数据,进行注释辅助双次比对,发现novel剪接事件,并与Illumina RNAseq交叉验证。
结果解读:(1)注释辅助方法在中等注释缺失(25%~66%)时效果最佳,剪接junction的true positive rate达0.86,precision达0.85(图7a-b);(2)hen2-2突变体中,过滤后的未注释junction有57%得到Illumina RNAseq支持,发现了AT1G19396(alternative donor位点导致的novel外显子)和AT3G12140(反义RNA的novel剪接)的剪接异构体(图8b-c)。
产品关联:使用STAR进行Illumina RNAseq比对,文献未提及具体商业产品。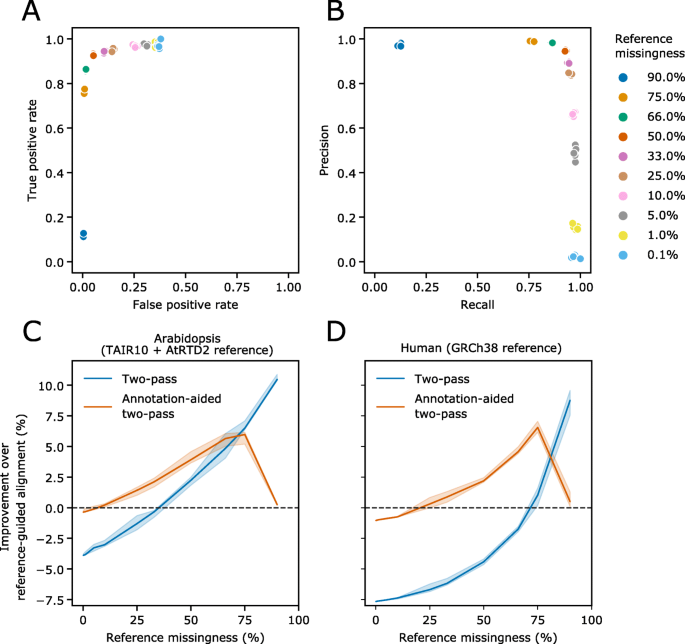
4. Biomarker研究及发现成果解析
本研究为方法学研究,聚焦于“准确的剪接junction”这一转录组特征的发现(广义上可视为转录组分析中的“Biomarker”)。
4.1 Biomarker定位
这里的“Biomarker”是长读长RNA测序中准确的剪接junction,筛选逻辑为“第一次比对提取→机器学习过滤(整合比对metrics与序列特征)→第二次比对验证”。
4.2 研究过程详述
- 来源:剪接junction来自长读长数据的第一次比对结果;
- 验证方法:① 模拟数据中与参考注释比较(金标准);② 真实数据中与Illumina RNAseq数据交叉验证;
- 特异性与敏感性:用F1分数衡量——模拟数据中Arabidopsis的F1=0.954,人类F1=0.957;真实数据中,过滤后的junction与Illumina RNAseq的重叠率达57%(未过滤时仅20%)。
4.3 核心成果
(1)方法性能提升:2passtools的剪接junction检测F1分数显著高于现有方法(如minimap2的单次比对、FLAIR的后比对校正);
(2)novel剪接事件发现:在注释不完整的hen2-2突变体中,发现了2382个受外切体调控的novel剪接junction,这些事件可能代表真实的功能转录本(如外切体降解的靶标);
(3)创新性:整合比对metrics与序列特征的机器学习模型,解决了长读长RNA测序中“无注释依赖”与“高准确性”的矛盾,为非模式生物或疾病样本的转录组分析提供了可靠工具。
综上,本研究开发的2passtools为长读长RNA测序中的内含子检测提供了更准确的解决方案,推动了全长转录本的研究,具有重要的方法学价值。