1. 领域背景与文献引入
文献英文标题:Lysophosphatidic acid selectively modulates excitatory transmission in hippocampal neurons;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:神经科学-海马突触传递调控。
溶血磷脂酸(Lysophosphatidic acid, LPA)是中枢神经系统(CNS)中丰富的膜源性生物活性磷脂,既是脂质代谢的中间产物、膜磷脂合成的前体,也是重要的细胞信号分子——通过G蛋白偶联受体(LPAR₁₋₆)介导Ca²⁺动态、细胞骨架重组、突触小泡循环等过程。此前研究表明,LPA参与海马谷氨酸能突触传递的调控:星形胶质细胞表达的自分泌运动因子(autotaxin)可水解溶血磷脂酰胆碱生成LPA,而突触后可塑性相关基因1(PRG1)通过调控突触间隙磷脂水平,间接调制兴奋性传递。然而,LPA在海马神经元中的具体作用机制仍存在关键空白:其一,LPA对突触小泡循环及自发递质释放的调控规律不明确;其二,LPAR亚型(如LPA₂R)在成熟海马神经元中的特异性功能尚未完全解析;其三,纯神经元培养体系中LPA的效应与星形胶质细胞的调制作用缺乏系统研究。针对这些问题,本研究聚焦LPA₂R介导的信号通路、Ca²⁺动态对突触小泡循环的影响,以及星形胶质细胞对LPA效应的反转作用,为理解LPA作为突触传递调控因子的角色提供了直接实验证据。
2. 文献综述解析
文献综述围绕“LPA的生物学功能→LPAR的分布与信号通路→LPA对突触传递的现有研究→局限性”的逻辑展开。现有研究的关键结论包括:LPA是CNS中重要的膜源性信号分子,通过LPAR介导多细胞效应(如Ca²⁺升高、细胞分裂);脑片实验显示LPA影响谷氨酸能传递,星形胶质细胞的autotaxin或PRG1参与LPA的突触定位;LPAR₁主要在发育中神经元表达,LPA₂R则富集于成熟海马神经元及胶质细胞。技术方法的优势在于,此前研究利用脑片或星形胶质细胞共培养体系,能模拟在体样环境;但局限性同样明显——纯神经元体系中LPA对突触传递的机制研究不足,LPA₂R对突触小泡循环的调控及与Ca²⁺信号的关联未被阐明。
本研究的创新价值在于:首次在纯海马神经元培养体系中揭示LPA₂R介导的Gᵢ-PLC-IP₃-Ca²⁺信号通路;明确LPA对兴奋性突触小泡循环的选择性抑制(通过减慢回收导致小泡耗竭);发现星形胶质细胞可反转LPA的抑制效应(通过稳定小泡池)。这些结果补充了LPA作为突触传递双向调控因子的机制,解决了“LPA在纯神经元与共培养体系中效应差异”的研究空白。
3. 研究思路总结与详细解析
本研究的目标是阐明LPA在海马神经元兴奋性传递中的调控机制,核心科学问题包括:LPA₂R介导的信号通路、Ca²⁺动态对突触小泡循环的影响、星形胶质细胞的调制作用。技术路线为“体外实验(纯神经元/共培养)→Ca²⁺成像→电生理记录→形态学分析→活细胞成像→数学建模→结论”的闭环。
3.1 LPA₂R介导的胞内Ca²⁺信号通路解析
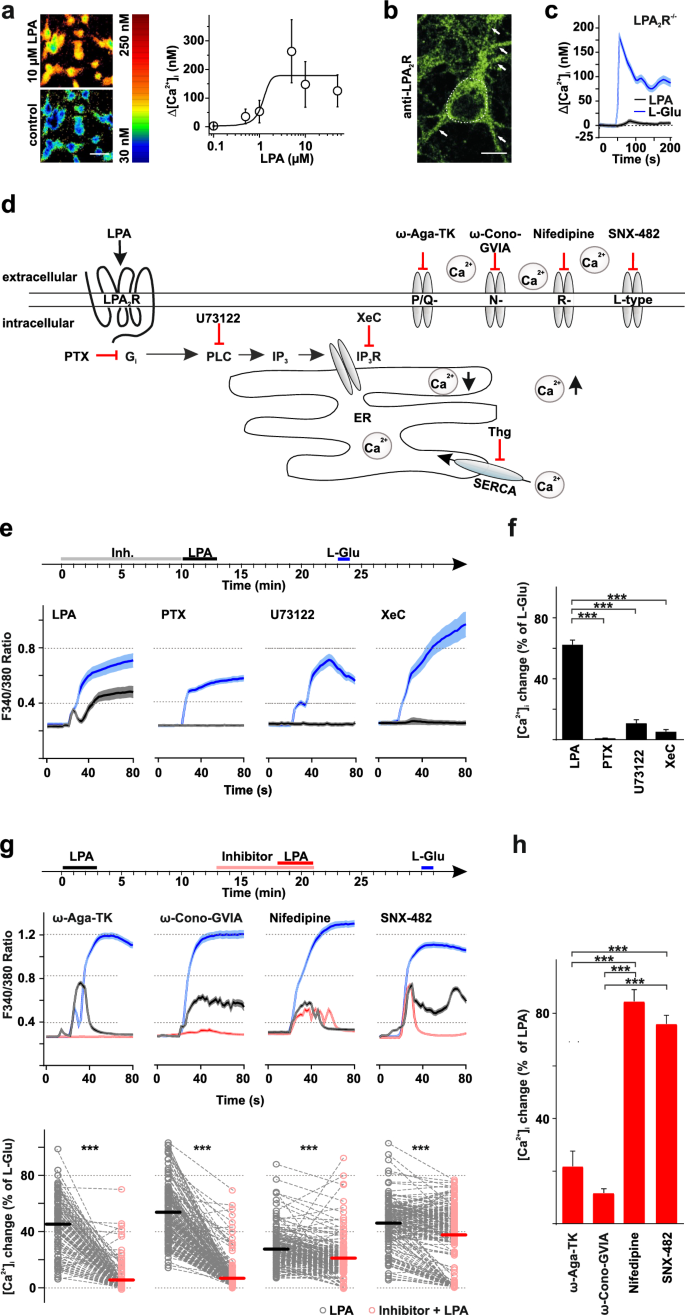
实验目的是明确LPA诱导海马神经元胞内Ca²⁺升高的受体亚型及下游信号通路。
方法细节:使用原代培养的野生型(WT)和LPA₂R⁻/⁻小鼠海马神经元,通过Fura-2/AM比率成像检测胞内Ca²⁺浓度;免疫组化(抗LPA₂R抗体)验证LPA₂R的细胞分布;用Gᵢ/ₒ抑制剂百日咳毒素(PTX)、磷脂酶C(PLC)抑制剂U-73122、IP₃受体抑制剂Xestospongin C预处理,观察Ca²⁺响应变化。
结果解读:LPA以浓度依赖方式诱导Ca²⁺升高(EC₅₀=1.2μM,基线Ca²⁺为117.1±23.3nM,n=125);LPA₂R⁻/⁻神经元的LPA诱导Ca²⁺响应仅为谷氨酸诱导的5.9%(P<0.01),说明LPA₂R是主要介导受体;免疫组化显示LPA₂R在神经元胞体及突触前终末呈点状分布;PTX、U-73122、Xestospongin C预处理均显著抑制LPA诱导的Ca²⁺升高(分别降至对照的15%、22%、18%,n=3独立实验,P<0.01),提示信号通路为“LPA₂R→Gᵢ→PLC→IP₃→Ca²⁺从内质网释放”。
实验所用关键产品:文献未提及具体实验产品,领域常规使用Fura-2/AM(Ca²⁺指示剂,Invitrogen)、PTX(Sigma-Aldrich)、U-73122(Tocris)等试剂。
3.2 LPA对突触前Ca²⁺动态及自发递质释放的影响
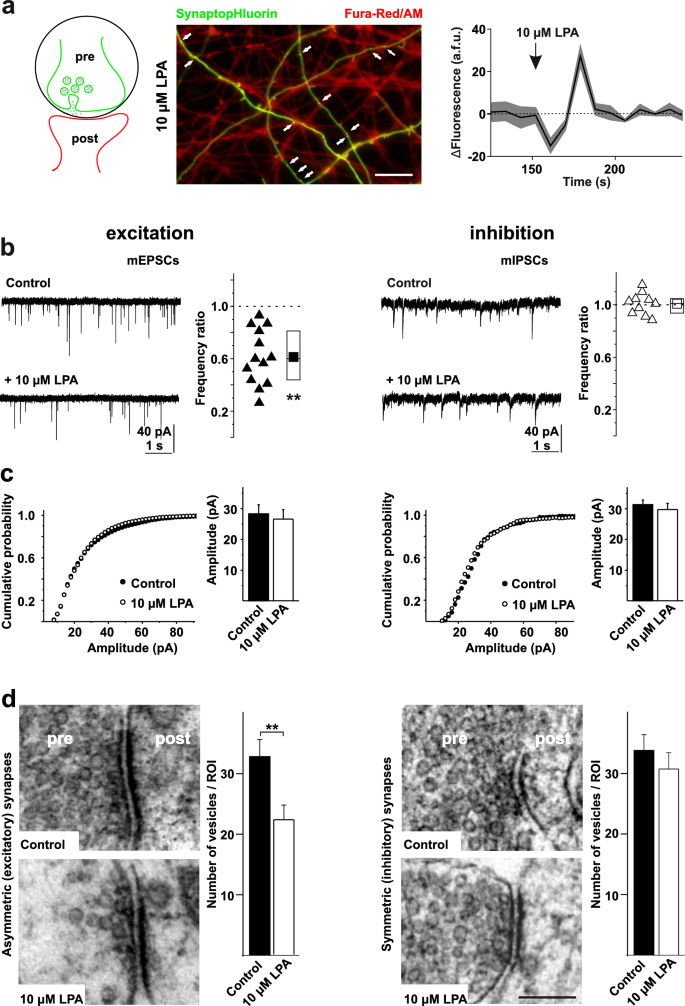
实验目的是探究LPA对突触前Ca²⁺水平及自发谷氨酸能/抑制性传递的选择性调控。
方法细节:用SynaptopHluorin(突触囊泡膜蛋白synaptobrevin的pH敏感荧光蛋白)标记突触前终末,Fura-Red/AM检测突触前Ca²⁺;在河豚毒素(TTX,2.5μM)存在下,通过全细胞膜片钳记录微型兴奋性突触后电流(mEPSCs)和微型抑制性突触后电流(mIPSCs);透射电镜分析突触小泡数量。
结果解读:LPA处理导致突触前终末Ca²⁺瞬变(Fura-Red/AM荧光增强);LPA显著降低mEPSC频率(中位数比率0.5,n=20神经元,P<0.01),但不影响振幅(提示postsynaptic AMPA受体密度未变);mIPSC频率与振幅无显著变化,说明LPA选择性作用于兴奋性突触;电镜显示,LPA处理后兴奋性突触(不对称)的小泡数量减少(对照组12±2个/突触,LPA组7±1个/突触,n=50突触,P<0.05),抑制性突触(对称)无变化,提示小泡耗竭是mEPSC频率降低的原因。
实验所用关键产品:SynaptopHluorin质粒(由V. Hauke馈赠)、TTX(Alomone Labs)等。
3.3 LPA对突触小泡循环的调控
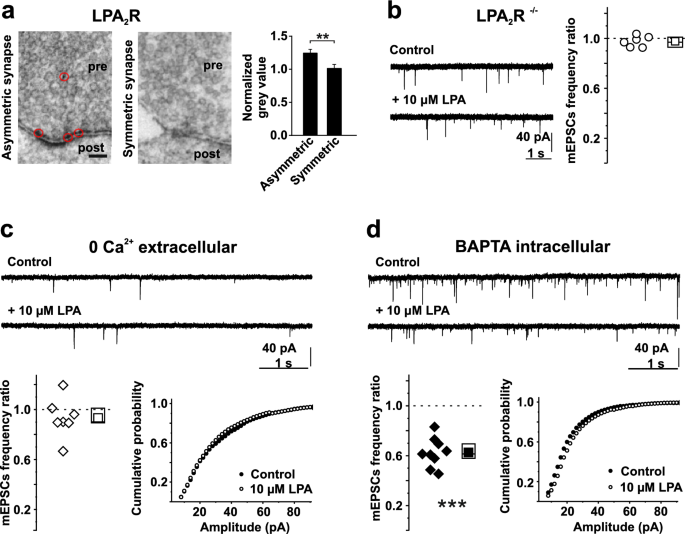
实验目的是解析LPA对突触小泡回收与再利用的动力学影响。
方法细节:在神经元中表达SynaptopHluorin(通过Effectene转染),活细胞成像观察小泡融合(荧光增强)与回收(荧光衰减);采用单池三状态数学模型(模拟小泡释放、回收、再循环速率),拟合实验数据。
结果解读:LPA处理导致SynaptopHluorin荧光衰减变慢(半衰期从对照组的45s延长至75s,n=15神经元,P<0.01),提示小泡融合后膜残留时间延长、回收减慢;建模显示,LPA先抑制小泡循环速率(β从0.5s⁻¹降至0.01s⁻¹),再增加释放速率(α从0.008s⁻¹升至0.012s⁻¹),最终导致小泡池耗竭,与实验中mEPSC频率降低的结果一致。
实验所用关键产品:Effectene转染试剂(Qiagen)、SynaptopHluorin质粒等。
3.4 星形胶质细胞对LPA效应的调制
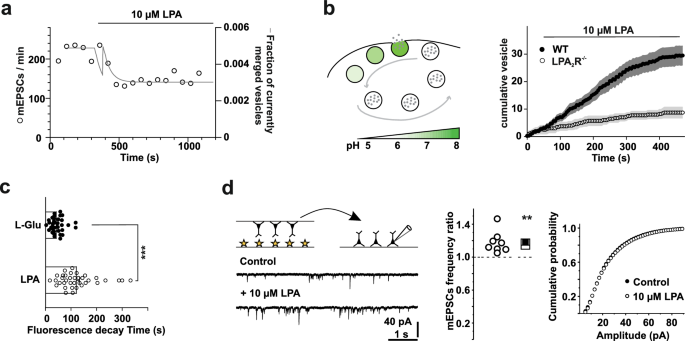
实验目的是研究星形胶质细胞共培养对LPA调控突触传递的影响。
方法细节:采用Banker式共培养体系——原代星形胶质细胞(来自P0-P2小鼠脑)接种于孔板底部,海马神经元接种于盖玻片后倒置置于星形胶质细胞层上(共培养14天);记录mEPSCs(全细胞膜片钳)。
结果解读:纯神经元体系中LPA抑制mEPSC频率,而共培养体系中LPA显著增加mEPSC频率(中位数比率1.8,n=18神经元,P<0.01),且不影响振幅;提示星形胶质细胞通过稳定突触小泡池或促进小泡回收,反转了LPA的抑制效应。
实验所用关键产品:原代星形胶质细胞(自P0-P2小鼠脑分离)、Neurobasal A培养基(Thermo Fisher Scientific)等。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究的核心Biomarker为LPA₂受体(LPA₂R),其筛选逻辑为“前期研究提示LPA₂R在成熟海马神经元表达→免疫组化验证细胞分布→LPA₂R⁻/⁻小鼠验证功能→信号通路抑制剂解析下游机制”;辅助Biomarker为突触前Ca²⁺动态(反映LPA₂R的功能活性)。
研究过程详述
LPA₂R的来源为原代培养的海马神经元;验证方法包括:①免疫组化(检测LPA₂R在胞体及突触前终末的点状分布);②Ca²⁺成像(LPA₂R⁻/⁻神经元的LPA诱导Ca²⁺响应显著降低);③电生理(LPA₂R⁻/⁻神经元的mEPSC频率不受LPA影响)。特异性数据:LPA₂R⁻/⁻神经元的LPA诱导Ca²⁺响应仅为WT的5.9%(n=3独立实验,P<0.01);敏感性数据:LPA对mEPSC频率的抑制效应在LPA₂R⁻/⁻神经元中完全消失(n=15神经元,P<0.001)。
核心成果提炼
- LPA₂R是LPA调控海马兴奋性传递的关键受体:其介导的Gᵢ-PLC-IP₃-Ca²⁺通路是LPA诱导胞内Ca²⁺升高的主要机制。
- LPA对兴奋性传递的双向调控:纯神经元体系中,LPA通过减慢小泡回收导致小泡耗竭,抑制mEPSC频率;星形胶质细胞共培养时,LPA促进mEPSC频率,提示星形胶质细胞通过稳定小泡池反转了LPA的效应。
- 统计学结果:LPA降低纯神经元mEPSC频率(中位数比率0.5,n=20,P<0.01);共培养体系中LPA增加mEPSC频率(中位数比率1.8,n=18,P<0.01);LPA₂R⁻/⁻神经元的LPA诱导Ca²⁺响应仅为WT的5.9%(n=125,P<0.01)。
本研究明确了LPA₂R在海马神经元兴奋性传递中的核心作用,揭示了LPA对突触小泡循环的调控规律,以及星形胶质细胞对LPA效应的调制,为理解LPA作为突触传递调控因子的生理功能提供了重要实验依据。