1. 领域背景与文献引入
文献英文标题:USP43 promotes cerebral ischemia–reperfusion injury via activation of TAK1;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:脑缺血再灌注损伤(神经科学细分领域)。
脑卒中是全球非传染性疾病中导致残疾和死亡的首要原因,其中缺血性卒中占新发卒中的约65%。目前,再血管化技术(如药物溶栓、血管内取栓)是缺血性卒中的主要临床治疗手段,但受限于狭窄的治疗时间窗(通常<4.5小时)及潜在的出血风险,患者预后仍不理想。此外,再血管化治疗不可避免地会引发脑缺血再灌注(I-R)损伤,其病理过程涉及神经炎症、细胞凋亡、氧化应激和兴奋性毒性等复杂机制。尽管针对这些靶点的药物研发已取得一定进展,但多数仍处于细胞或动物实验阶段,缺乏有效的临床转化,因此亟需深入解析脑I-R损伤的分子机制,挖掘潜在治疗靶点。
泛素特异性蛋白酶43(USP43)作为去泛素化酶(DUB)家族成员,现有研究主要集中在肿瘤领域:其在乳腺癌、结直肠癌、骨肉瘤等肿瘤组织中上调,通过去泛素化稳定ZEB1、c-Myc等靶蛋白,促进肿瘤细胞增殖、转移及耐药。然而,USP43在神经科学领域的功能尚未明确,尤其在脑I-R损伤中的作用及机制尚无报道,这是当前领域的研究空白。
本研究旨在探讨USP43在脑I-R损伤中的作用及分子机制,其学术价值在于首次揭示USP43通过激活转化生长因子β激活激酶1(TAK1)及其下游c-Jun N端激酶(JNK)/p38通路,促进神经炎症和神经元凋亡的病理过程,为急性卒中的治疗提供了新的潜在靶点。
2. 文献综述解析
本文综述部分围绕“脑I-R损伤的临床挑战与病理机制”“USP43的结构与现有研究”及“研究空白”展开评述,逻辑清晰地衔接了研究背景与科学问题。
现有研究总结:①脑I-R损伤的临床现状:再血管化治疗是缺血性卒中的主要手段,但时间窗限制和再灌注损伤导致预后不佳;病理机制方面,神经炎症(如核因子κB(NF-κB)通路激活)和细胞凋亡(如Bcl2相关X蛋白(Bax)/B细胞淋巴瘤-2(Bcl2)失衡)是关键驱动因素。②USP43的结构与表达:USP43是含1123个氨基酸的线性蛋白,定位于人类染色体7p13.1,主要在脑、主动脉、肺等组织表达,脑内以神经元表达为主。③USP43的现有功能:在肿瘤领域,USP43通过去泛素化调控靶蛋白稳定性,促进肿瘤增殖、转移;在免疫调节、妊娠糖尿病、颈椎病等领域也有少量报道,但未涉及神经疾病。
现有研究的局限性:USP43在神经科学领域的功能完全未知,尤其缺乏其在脑I-R损伤中的作用研究。本研究的创新价值:首次将USP43与脑I-R损伤关联,通过体内外模型验证其促损伤功能,并解析了USP43-TAK1-JNK/p38的分子机制,填补了USP43在神经领域的研究空白。
3. 研究思路总结与详细解析
本研究采用“体内动物模型+体外细胞模型”的双向验证策略,围绕“USP43表达变化→功能验证→机制解析→抑制剂验证”的逻辑展开,完整回答了“USP43是否参与脑I-R损伤”“如何参与”及“机制是什么”的科学问题。
3.1 脑I-R模型构建与USP43表达验证
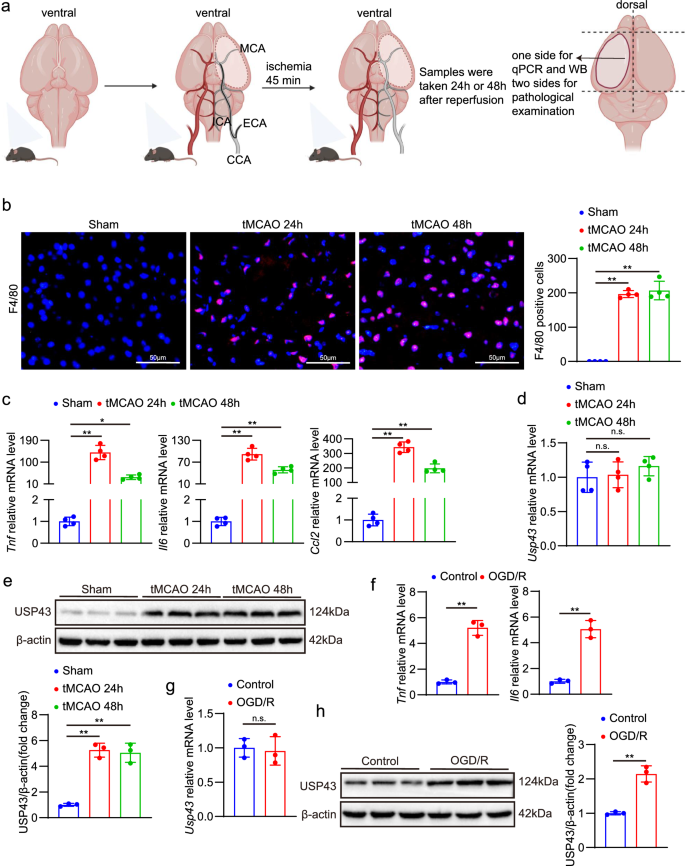
实验目的:构建小鼠脑I-R(短暂性大脑中动脉阻塞,tMCAO)模型和原代神经元缺氧复氧(OGD/R)模型,检测USP43的表达变化。
方法细节:①小鼠tMCAO模型:采用线栓法阻塞大脑中动脉45分钟,再灌注24或48小时后取脑样本;②原代大鼠皮层神经元OGD/R模型:神经元培养7天后,用无糖无血清培养基处理,置于95%氮气(N₂)+5%二氧化碳(CO₂)环境3小时(缺氧缺糖),再换正常培养基在常氧环境培养6小时(复氧);③通过实时荧光定量PCR(qPCR)检测USP43的mRNA水平,蛋白质印迹法(Western blot)检测蛋白水平。
结果解读:①体内模型:tMCAO处理后,小鼠脑内USP43蛋白水平在24、48小时显著升高(P<0.05或P<0.01,n=4),但mRNA水平无变化;②体外模型:OGD/R处理后,神经元USP43蛋白水平显著升高(P<0.01,n=3),mRNA水平无变化。这表明USP43在脑I-R损伤中通过转录后调控(如蛋白稳定性)发挥作用。
实验所用关键产品:线栓(Doccol,货号602156PK10Re)、Trizol试剂(用于RNA提取)、Western blot相关试剂(RIPA裂解液、蛋白酶抑制剂(Roche,货号0469312001)、BCA蛋白定量试剂盒(Thermo,货号23225))。
3.2 USP43基因敲除对脑I-R损伤的体内保护作用
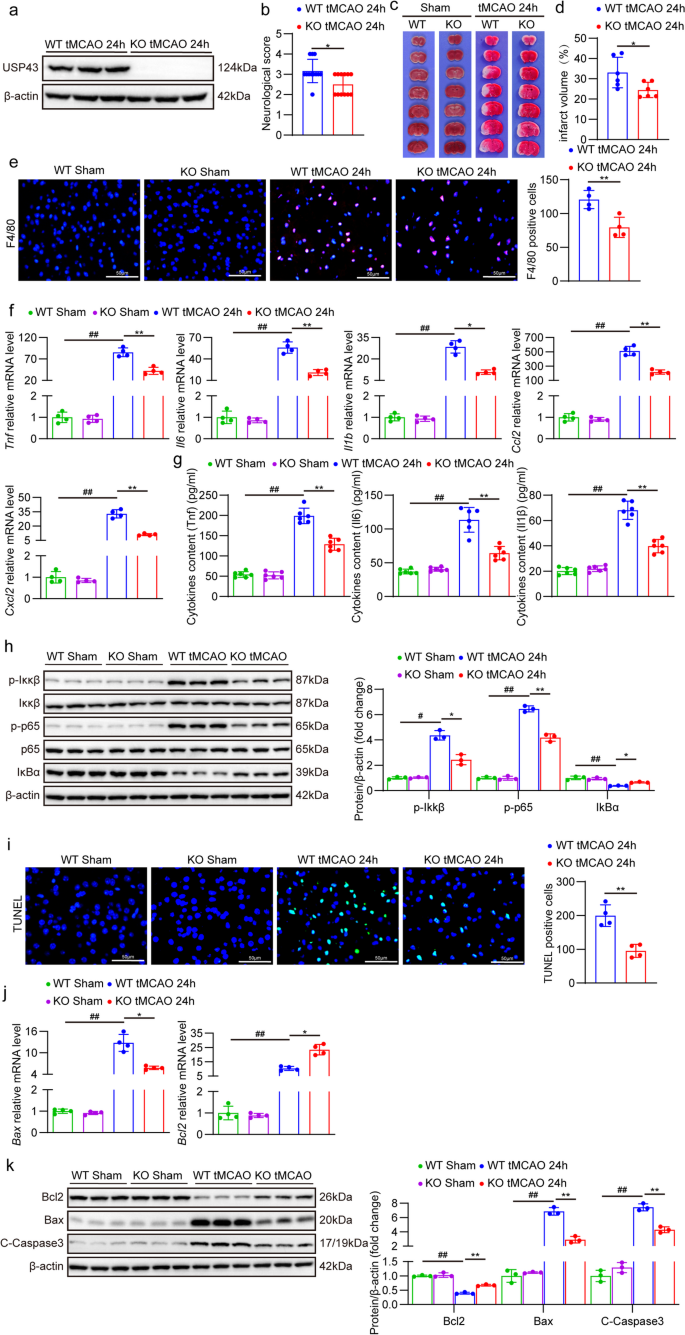
实验目的:通过USP43敲除小鼠,研究USP43缺失对脑I-R损伤的影响。
方法细节:①构建USP43敲除(USP43-KO)小鼠,通过Western blot验证敲除效率;②对野生型(WT)和USP43-KO小鼠进行tMCAO手术,24小时后评估:神经功能评分(改良9分法)、脑梗死体积(2,3,5-三苯基四唑氯化物(TTC)染色)、神经炎症(F4/80免疫荧光检测巨噬细胞/小胶质细胞浸润,qPCR检测炎症因子mRNA水平,酶联免疫吸附试验(ELISA)检测血清炎症因子)、细胞凋亡(末端脱氧核苷酸转移酶dUTP缺口末端标记(TUNEL)染色,qPCR/Western blot检测凋亡相关分子)、信号通路(Western blot检测NF-κB通路分子)。
结果解读:①神经功能与梗死体积:USP43-KO小鼠神经功能评分显著降低(P<0.01,n=12),脑梗死体积减小(P<0.01,n=6);②炎症反应:F4/80阳性细胞数减少(P<0.01,n=4),脑内肿瘤坏死因子(Tnf)、白细胞介素6(Il6)、C-C基序趋化因子配体2(Ccl2)等炎症因子mRNA水平降低(P<0.01,n=4),血清TNF-α、IL-6水平降低(P<0.01,n=6),NF-κB通路激活减弱(磷酸化的核因子κB抑制激酶β(p-Ikkβ)、磷酸化的核因子κB p65(p-p65)降低,核因子κB抑制蛋白α(IκBα)升高,P<0.01,n=3);③细胞凋亡:TUNEL阳性细胞数减少(P<0.01,n=4),Bax mRNA和蛋白水平降低,Bcl2升高(P<0.01,n=3/4)。
实验所用关键产品:USP43敲除小鼠(Ganan Institute of Innovation and Translational Medicine)、TTC染色试剂盒、F4/80抗体(Servicebio,货号GB11027)、ELISA试剂盒(ELK,如TNF-α货号ELK1387-1)。
3.3 USP43 knockdown对神经元OGD/R损伤的保护作用
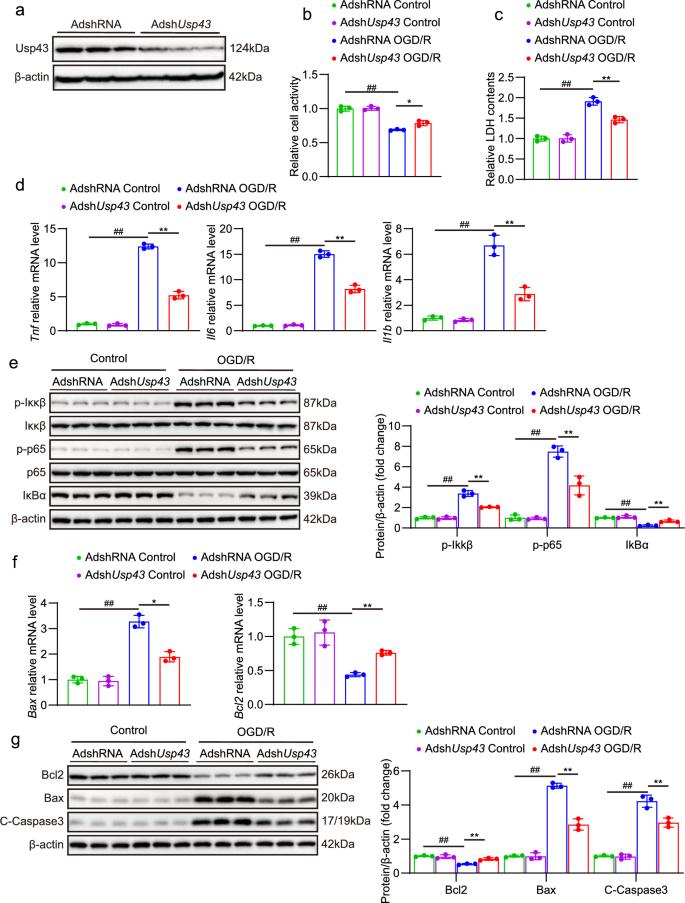
实验目的:在原代神经元中验证USP43缺失对OGD/R损伤的保护作用。
方法细节:①用USP43 knockdown腺病毒(AdshUsp43)感染原代神经元,Western blot验证敲除效率;②OGD/R处理后,检测细胞活力(细胞计数 kit-8(CCK-8))、乳酸脱氢酶(LDH)释放、炎症因子mRNA水平、NF-κB通路分子及凋亡相关分子。
结果解读:①细胞活力与损伤:AdshUsp43组细胞活力显著高于对照组(P<0.01,n=3),LDH释放减少(P<0.01,n=3);②炎症反应:Tnf、Il6、白细胞介素1β(Il1b)mRNA水平降低(P<0.01,n=4),NF-κB通路激活减弱(p-Ikkβ、p-p65降低,P<0.01,n=3);③细胞凋亡:Bax mRNA和蛋白水平降低,Bcl2升高(P<0.01,n=3/4)。
实验所用关键产品:AdshUsp43腺病毒、CCK-8试剂盒(Bicentennial,货号C0039)、LDH试剂盒(Bicentennial,货号C0016)。
3.4 USP43过表达对神经元OGD/R损伤的加重作用
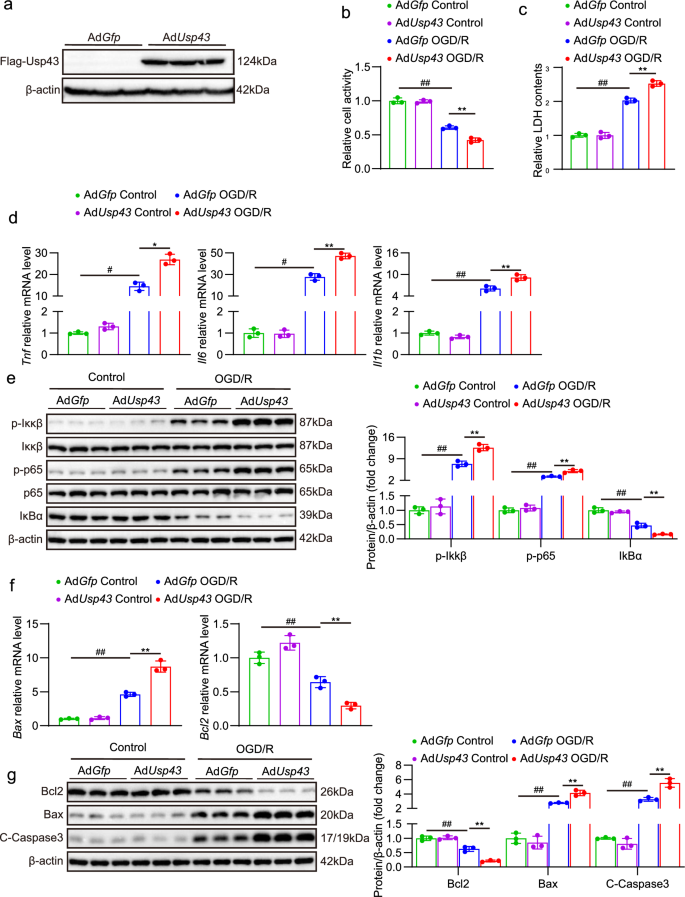
实验目的:验证USP43过表达对神经元OGD/R损伤的促损伤作用。
方法细节:①用USP43过表达腺病毒(AdUsp43)感染原代神经元,Western blot验证过表达效率;②OGD/R处理后,检测细胞活力、LDH释放、炎症因子、信号通路及凋亡分子。
结果解读:①细胞活力与损伤:AdUsp43组细胞活力降低(P<0.01,n=3),LDH释放增加(P<0.01,n=3);②炎症反应:Tnf、Il6、Il1b mRNA水平升高(P<0.01,n=4),NF-κB通路激活增强(p-Ikkβ、p-p65升高,P<0.01,n=3);③细胞凋亡:Bax mRNA和蛋白水平升高,Bcl2降低(P<0.01,n=3/4)。
实验所用关键产品:AdUsp43腺病毒。
3.5 USP43对TAK1-JNK/p38通路的调控
实验目的:解析USP43调控脑I-R损伤的信号通路。
方法细节:①体内:检测WT和USP43-KO小鼠脑内丝裂原活化蛋白激酶(MAPK)通路分子(细胞外调节蛋白激酶(ERK)、JNK、p38)及上游TAK1、凋亡信号调节激酶1(ASK1)的磷酸化水平;②体外:检测AdshUsp43/AdUsp43感染神经元中上述分子的磷酸化水平。
结果解读:①体内:tMCAO处理后,WT小鼠脑内ERK、JNK、p38磷酸化水平升高;USP43-KO小鼠JNK、p38磷酸化显著降低(P<0.01,n=3),ERK无变化;TAK1磷酸化降低(P<0.01,n=3),ASK1无变化。②体外:AdshUsp43组JNK、p38磷酸化降低(P<0.01,n=3),AdUsp43组升高(P<0.01,n=3);TAK1磷酸化变化趋势一致。
实验所用关键产品:Western blot抗体(如p-JNK、p-p38、TAK1,品牌如Cell Signaling Technology)。
3.6 TAK1抑制剂对USP43过表达的逆转作用
实验目的:验证USP43的促损伤作用依赖TAK1激活。
方法细节:①用TAK1抑制剂5Z-7-oxozeaenol(600 nM)预处理AdUsp43感染的神经元1小时;②OGD/R处理后,检测TAK1、JNK、p38磷酸化水平,细胞活力、LDH释放、炎症因子及凋亡分子。
结果解读:①信号通路:抑制剂处理后,TAK1、JNK、p38磷酸化水平降低(P<0.01,n=3);②细胞活力与损伤:细胞活力升高(P<0.01,n=3),LDH释放减少(P<0.01,n=4);③炎症与凋亡:Tnf、Il6、Il1b mRNA水平降低(P<0.01,n=4),Bax mRNA和蛋白水平降低,Bcl2升高(P<0.01,n=3/4)。
实验所用关键产品:TAK1抑制剂(Sigma,货号O9890-1 MG)。
3.7 USP43与TAK1的相互作用及泛素化调控
实验目的:揭示USP43调控TAK1的分子机制。
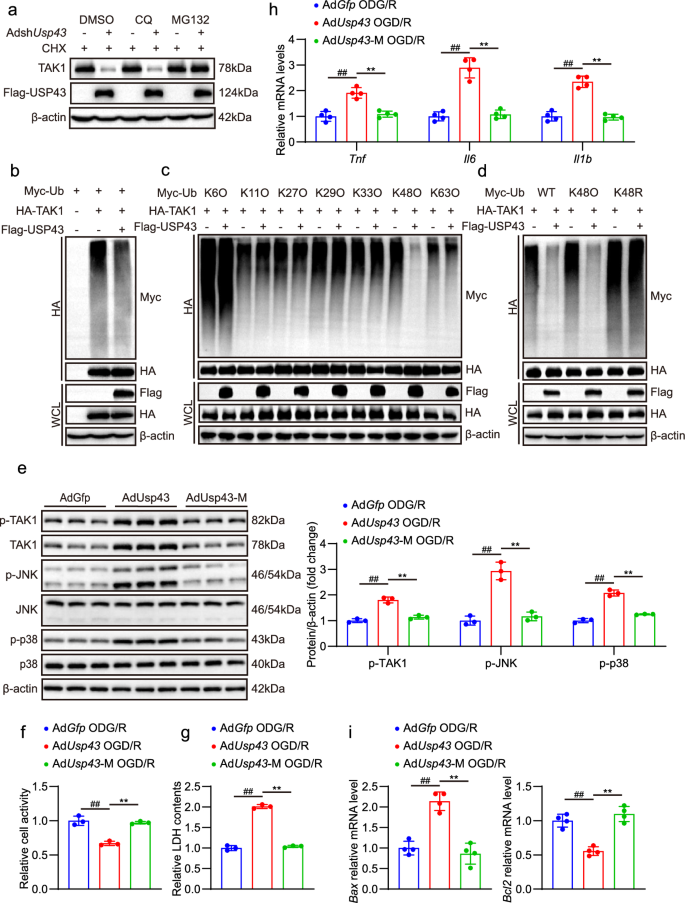
方法细节:①免疫共沉淀(IP):人胚肾293T(HEK293T)细胞共转染Flag-USP43和HA-TAK1,检测相互作用;②谷胱甘肽S-转移酶(GST)pull-down:验证USP43与TAK1的直接相互作用;③截短体实验:构建TAK1(1-300、301-600、601-706aa)和USP43(1-712、713-1123aa)截短体,检测结合结构域;④环己酰亚胺(CHX)实验:用蛋白合成抑制剂CHX处理AdUsp43感染的神经元,检测TAK1稳定性;⑤泛素化实验:共转染Flag-USP43、HA-TAK1和Myc-泛素(或K48/K63特异性泛素),检测TAK1的泛素化水平。
结果解读:①相互作用:USP43与TAK1在HEK293T细胞和原代神经元中均存在相互作用,且为直接结合;②结合结构域:TAK1的N端1-300aa是结合必需区域,USP43的1-712和713-1123aa均能结合TAK1;③蛋白稳定性:AdUsp43组TAK1蛋白半衰期延长(CHX处理12小时后仍有较高水平,P<0.01,n=3);④泛素化调控:USP43降低TAK1的总泛素化水平,尤其显著去除K48链泛素化(P<0.01,n=3);蛋白酶体抑制剂MG132可恢复AdshUsp43组TAK1的降解(P<0.01,n=3),而溶酶体抑制剂氯喹(CQ)无作用。
实验所用关键产品:转染试剂PEI(Polysciences,货号24765-100)、GST beads、Myc-泛素质粒、CHX、MG132(Roche,货号0469312001)、CQ(Sigma)。
4. Biomarker研究及发现成果解析
本文中USP43作为脑I-R损伤的功能性生物标志物(Biomarker),通过“疾病模型表达上调→功能验证→机制解析”的完整链条,明确了其作为促损伤Biomarker的价值。
Biomarker定位与筛选逻辑
USP43是脑I-R损伤的蛋白质类Biomarker,筛选逻辑为:①疾病模型中表达上调:tMCAO/OGD/R模型中USP43蛋白水平显著升高;②功能验证:基因敲除/knockdown后损伤减轻,过表达后损伤加重;③机制解析:通过相互作用、泛素化及信号通路实验,明确USP43通过调控TAK1通路发挥作用。
研究过程详述
①Biomarker来源:脑I-R损伤模型的脑组织(小鼠tMCAO)和原代神经元(OGD/R);②验证方法:qPCR和Western blot检测表达水平,IP检测相互作用,CHX实验检测稳定性,泛素化实验检测修饰机制,抑制剂实验验证功能依赖;③特异性与敏感性:在tMCAO模型中,USP43蛋白水平在24/48小时显著升高(P<0.05或P<0.01,n=4);在OGD/R神经元中,USP43蛋白水平升高(P<0.01,n=3);④功能关联:USP43与脑I-R损伤的严重程度正相关(敲除后损伤减轻,过表达后加重),神经功能评分和梗死体积的统计学差异显著(P<0.01)。
核心成果提炼
①功能性:USP43通过去泛素化稳定TAK1,激活JNK/p38通路,促进神经炎症和神经元凋亡,是脑I-R损伤的促损伤Biomarker;②创新性:首次发现USP43在脑I-R损伤中的作用,揭示其与TAK1的相互作用及K48链泛素化调控机制;③统计学结果:USP43敲除小鼠梗死体积减小(P<0.01,n=6),神经元knockdown后细胞活力升高(P<0.01,n=3),过表达后LDH释放增加(P<0.01,n=3)。
综上,本研究首次揭示了USP43在脑I-R损伤中的促损伤作用及分子机制,为急性卒中的治疗提供了新的潜在靶点。