1. 领域背景与文献引入
文献英文标题:Evaluating nanopore sequencing data processing pipelines for structural variation identification;发表期刊:Genome Biology;影响因子:10.806(2019年);研究领域:基因组学-结构变异检测-纳米孔测序数据处理。
结构变异(Structural Variation, SV)是基因组中大于50bp的 DNA 片段改变(包括插入、缺失、重复、倒位、易位),占人类个体基因组差异的90%以上,与癌症、神经疾病等重大疾病的发生发展密切相关。二代测序(Next-Generation Sequencing, NGS)因短读长(<200bp)限制,需依赖读深、配对读错配等间接证据检测 SV,准确性低且易漏检重复区域的变异。三代纳米孔测序(Oxford Nanopore Technologies, ONT)可生成超长读长(可达 Mb 级),直接覆盖 SV 区域,理论上能显著提高 SV 检测性能。但截至2019年,针对纳米孔长读长的比对工具(Aligner)和 SV 检测工具(Caller)尚未被系统评估,不同工具组合的性能差异、测序深度的影响及多流程整合方法均未明确,成为纳米孔测序应用于 SV 研究的关键瓶颈。
本文针对这一空白,系统评估了4种比对工具(minimap2、NGMLR、GraphMap、LAST)与3种 SV 检测工具(Sniffles、NanoSV、Picky)的组合性能,分析测序深度对 SV 检测的影响,并开发机器学习模型整合多个流程的 call set,为纳米孔测序 SV 检测提供了标准化 workflow。
2. 文献综述解析
作者对现有研究的分类维度为“测序技术-工具类型-性能局限”:
1. 二代测序的 SV 检测局限:短读长导致间接证据依赖,重复区域、大尺寸 SV(>1kb)的检测准确性极低,假阳性率高达30%-50%;
2. 三代测序的工具现状:纳米孔、PacBio 等长读长测序技术已成熟,但配套的比对工具(如 minimap2、NGMLR)和 SV 检测工具(如 Sniffles、NanoSV)均为近年开发,缺乏跨数据集、跨工具的系统比较;
3. 现有整合方法的不足:部分研究尝试合并多个工具的 call set,但多采用“简单共识”(如要求≥2个工具支持),未利用 SV 的特征信息(如长度、支持读长数)优化,性能提升有限。
现有研究的核心局限是工具评估的碎片化——多数研究仅用单一数据集或工具组合,无法反映真实场景下的性能差异;而本文的创新点在于:
- 首次在4个数据集(2个真实纳米孔数据、2个模拟数据)上系统比较7种工具组合的性能;
- 量化测序深度对 SV 检测的影响,明确20×为最低有效覆盖度;
- 开发随机森林模型整合多个流程的特征(如 SV 长度、支持读长数),显著提升 call set 准确性(F1 从0.47提高到0.79)。
3. 研究思路总结与详细解析
本文的研究目标是建立纳米孔测序 SV 检测的标准化 workflow,核心科学问题是“哪些工具组合能高效准确检测 SV?如何整合多流程提高性能?”,技术路线为“数据集生成→Read Mapping→SV 鉴定→性能评估→覆盖度分析→共识 call set→机器学习整合”。
3.1 数据集生成
实验目的:构建“真实+模拟”的多样化数据集,全面测试工具在不同场景下的性能。
方法细节:
- 真实数据集:选取 NA12878(~30×纳米孔测序,GIAB 提供 PacBio 真实 SV 集)、CHM13(~50×纳米孔测序,dbVar 提供 PacBio 真实 SV 集);
- 模拟数据集:用 NanoSim 模拟 CHM1 基因组的纳米孔读长(~50×覆盖度,Eichler Lab 提供 PacBio 真实 SV 集);用 RSVSim 在 GRCh38 20号染色体人工引入 SV(与 NA12878 真实集的大小、数量一致),生成~50×覆盖度的模拟读长。
结果解读:真实数据集反映临床样本的测序特征(如读长分布、错误率),模拟数据集(尤其是 Chr20)可精准计算工具的真阳性/假阳性率,解决真实集“SV 不完全”的问题。
产品关联:实验所用关键工具:NanoSim(纳米孔读长模拟,ver 2.1.0)、RSVSim(SV 模拟,ver 1.24.0)、liftOver(基因组版本转换)。
3.2 Read Mapping 与 SV 鉴定
实验目的:比较4种比对工具与3种 SV 检测工具的组合性能。
方法细节:
- 比对工具:测试 minimap2(快速长读长比对)、NGMLR(针对 SV 优化的比对)、GraphMap(敏感比对)、LAST(传统长读长比对);
- SV 检测工具:测试 Sniffles(快速 SV 检测,支持 SAM 格式)、NanoSV(基于读深的 SV 检测)、Picky(与 LAST 协同,支持 MAF 格式);
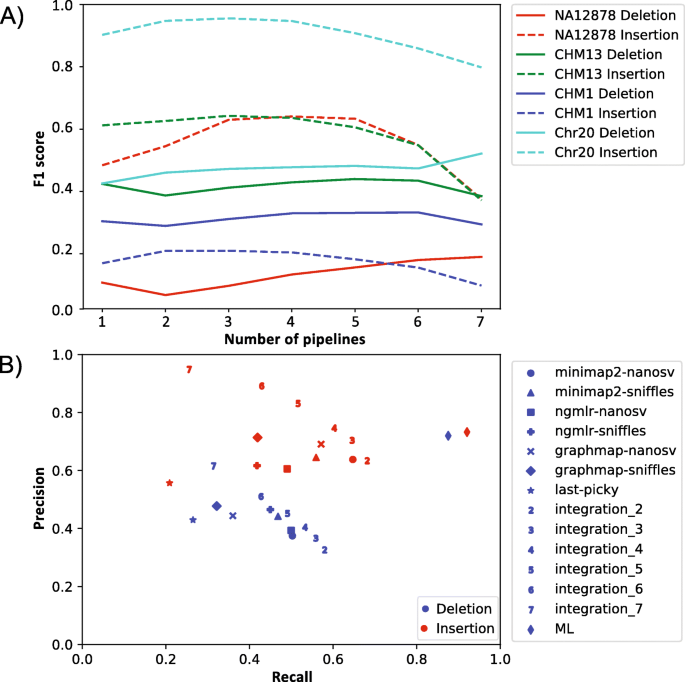
- 组合方式:共7种流程:Minimap2-NanoSV、NGMLR-NanoSV、GraphMap-NanoSV、Minimap2-Sniffles、NGMLR-Sniffles、GraphMap-Sniffles、LAST-Picky。
结果解读:
- 比对工具性能:minimap2 是最快的工具(比 GraphMap 快4-5倍),且映射的碱基数最多(比 LAST 多15%);
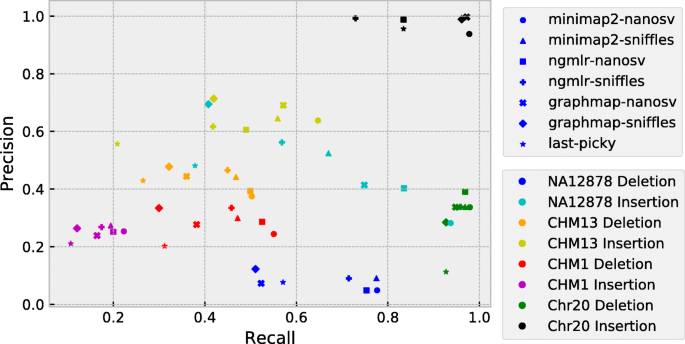
- SV 检测工具性能:NanoSV 的召回率(Recall)最高(NA12878 插入的 Recall=85%),但精确率(Precision)最低(70%);Sniffles 的精确率最高(90%),召回率中等(80%);Picky 的 F1 分数最低(0.65)。
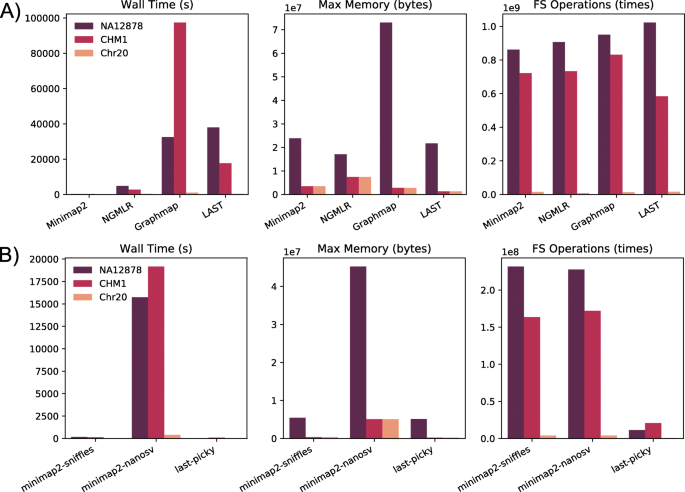
产品关联:实验所用关键工具:minimap2(ver 2.17)、NGMLR(ver 0.2.7)、Sniffles(ver 1.0.11)、NanoSV(ver 1.2.4)、SAMtools(ver 1.6,映射统计)。
3.3 Call Set 评估
实验目的:量化不同流程的 SV 检测性能(Precision、Recall、F1)。
方法细节:
- 用 BEDTools 比较 call set 与真实集:缺失需≥50%区域重叠,插入需≤100bp 距离;
- 计算 Precision(真阳性/总阳性)、Recall(真阳性/真总数)、F1(2(Precision×Recall)/(Precision+Recall))。
结果解读:
- 流程性能因 SV 类型而异:Minimap2-NanoSV 适合高召回率需求(如癌症样本的 SV 筛查),GraphMap-Sniffles 适合高精确率需求(如临床诊断);
- 模拟数据(Chr20)的插入检测性能最好(F1=0.92),真实数据(CHM1)的插入检测性能最差(F1=0.45),因 CHM1 基因组的重复区域更多。
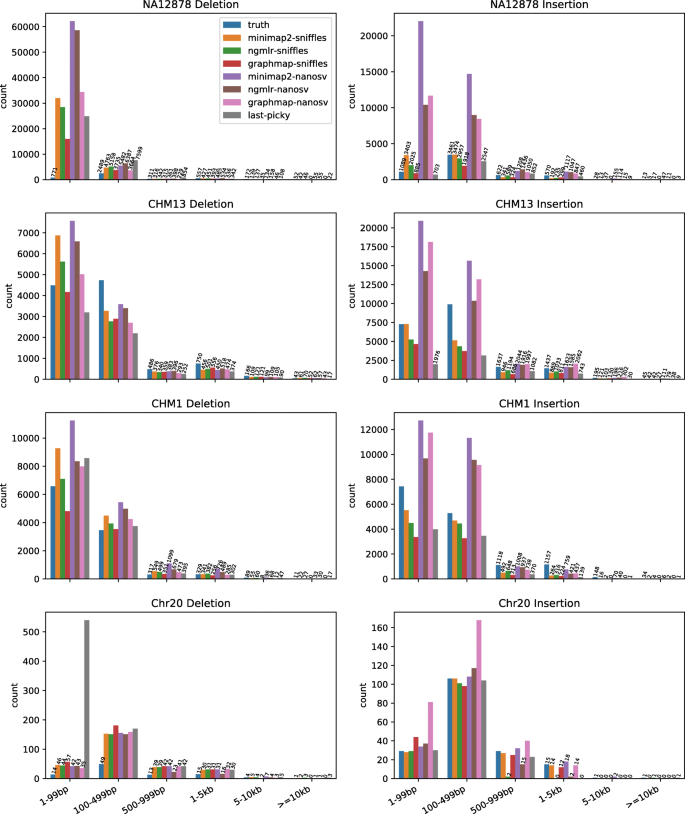
产品关联*:实验所用关键工具:BEDTools(ver 2.27.1,call set 比较)、matplotlib(绘图)。
3.4 覆盖度分析
实验目的:明确纳米孔 SV 检测的最低有效覆盖度。
方法细节:用 seqtk 对原始数据亚采样,生成10×、20×、30×、40×、50×覆盖度的数据集,用 Minimap2-Sniffles 流程检测 SV,计算 F1。
结果解读:
- 覆盖度≥20×时,F1 保持稳定(如 NA12878 缺失的 F1=0.8);
- 覆盖度<20×时,F1 急剧下降(10×覆盖度的 F1=0.5,n=3次重复,P<0.01)。
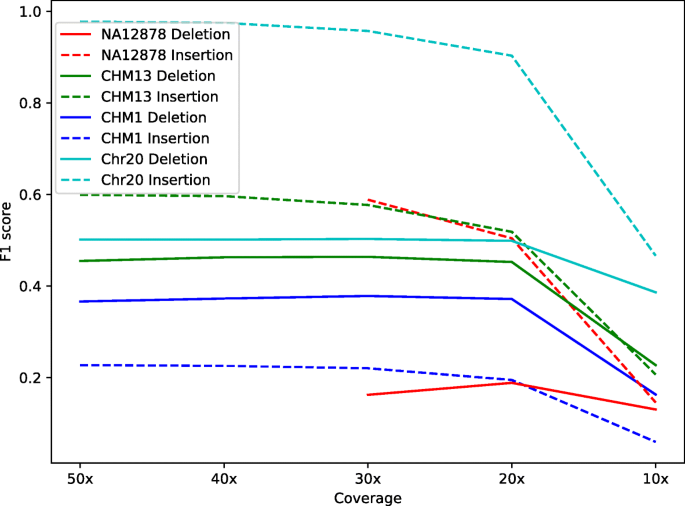
产品关联:实验所用关键工具:seqtk(亚采样)、minimap2(比对)、Sniffles(SV 检测)。
3.5 共识 Call Set 生成
实验目的:测试整合多个流程对性能的提升。
方法细节:合并所有7个流程的 call set,统计每个 SV 被多少个流程支持,过滤需要≥2/3/…/7个流程支持的 call set,计算 F1。
结果解读:
- 缺失需6-7个流程支持时 F1 最高(如 NA12878 缺失的 F1 从0.75提高到0.85);
- 插入需2-3个流程支持时 F1 最高(如 CHM13 插入的 F1 从0.65提高到0.78)。
3.6 机器学习预测
实验目的:利用 SV 特征优化整合流程,进一步提升性能。
方法细节:
- 提取每个 SV 的7个特征:长度、支持读长数、映射质量、深度 P 值、断点置信区间等;
- 用 CHM13 数据集训练随机森林模型(XGBoost),预测 SV 的真实性(真阳性/假阳性)。
结果解读:
- 机器学习模型的 F1 显著高于简单共识(CHM13 缺失的 F1 从0.47提高到0.79,插入从0.67提高到0.81);
- 最重要的特征是 SV 长度(贡献~50%),其次是深度 P 值(贡献~20%)。
产品关联:实验所用关键工具:XGBoost(ver 0.90,随机森林)、scikit-learn(ver 0.21.3,数据分割/评估)。
4. Biomarker 研究及发现成果解析
本文的核心是纳米孔测序 SV 检测的工具评估,而非鉴定特定的 Biomarker,但为 SV 作为疾病 Biomarker 的研究提供了关键技术支撑:
4.1 Biomarker 定位与筛选逻辑
文献未聚焦特定 SV 作为 Biomarker,而是建立了可靠的 SV 检测 workflow——通过系统评估工具组合,明确 Minimap2-Sniffles 为初始分析的首选流程(快速且性能平衡),整合多个流程的机器学习方法为深度分析的最优方案(准确性最高)。这些结果为后续研究(如癌症样本的 SV Biomarker 筛查)提供了标准化技术路径。
4.2 核心成果与学术价值
- 技术层面:明确纳米孔 SV 检测的最低覆盖度(20×)、最优工具组合(Minimap2-Sniffles)及整合方法(随机森林),解决了“如何用纳米孔测序准确检测 SV”的关键问题;
- 应用层面:为 SV 作为疾病 Biomarker 的研究提供了可靠的技术基础——例如,癌症样本的 SV 检测可采用 Minimap2-NanoSV 高召回率流程筛查候选 Biomarker,再用机器学习模型过滤假阳性,最终得到高置信的 SV Biomarker。
本文通过系统的工具评估和流程优化,为纳米孔测序在 SV 研究中的应用提供了标准化指南,推动了长读长测序技术在基因组学研究中的转化应用。