1. 领域背景与文献引入
文献英文标题:Human papillomavirus 18 E6 inhibits phosphorylation of p53 expressed in HeLa cells;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:肿瘤学-宫颈癌与人乳头瘤病毒(HPV)致癌机制。
领域共识:全球每年约47万宫颈癌新发病例,23万患者死亡,其中80%集中在发展中国家,HPV感染是宫颈癌的主要致病因素,99.7%的宫颈癌组织中可检测到整合的HPV DNA,HPV16和18型是高危型别,占宫颈癌病例的50%以上。HPV的E6和E7癌蛋白是核心致癌因子,已知E6可通过泛素依赖或非依赖的蛋白酶体途径降解p53,还能结合并降解乙酰转移酶p300抑制p53乙酰化,同时降解下游凋亡诱导因子bax,从而完全失活p53的抑癌功能。当前研究热点聚焦于HPV致癌的分子机制与靶向干预,但核心未解决问题是:在HPV阳性细胞中,过表达的野生型p53为何无法发挥抑癌功能,现有研究仅关注降解或乙酰化调控,未涉及磷酸化对p53激活的关键作用。针对这一空白,本研究旨在明确E6是否通过抑制p53磷酸化而非降解途径失活其功能,为HPV相关肿瘤的治疗提供新的机制依据。
2. 文献综述解析
作者对领域内现有研究的分类维度为E6失活p53的分子机制类型,包括降解途径调控、乙酰化修饰抑制、下游因子降解三个方向。现有研究的关键结论为:E6通过与E6AP形成复合物介导p53泛素化降解,通过结合p300抑制p53乙酰化,通过降解bax阻断p53下游凋亡通路;技术方法优势在于明确了E6与p53相互作用的核心分子复合物,为HPV致癌机制提供了直接证据;局限性在于所有研究均聚焦于p53的降解或乙酰化调控,未关注磷酸化修饰对p53激活的必要性,尤其是在HPV阳性细胞中过表达p53的功能失活机制尚未阐明,无法解释过表达p53无法抑制肿瘤细胞生长的现象。本研究的创新价值在于首次揭示了E6通过抑制p53磷酸化而非降解途径失活其功能,补充了E6调控p53的新机制,为HPV相关肿瘤中p53功能的恢复提供了新的靶点方向。
3. 研究思路总结与详细解析
本研究的核心科学问题是:HPV18 E6是否通过抑制p53磷酸化而非降解途径,导致HPV阳性细胞中过表达的p53功能失活;研究目标是明确E6调控p53的新机制;技术路线为:构建DOX诱导的p53过表达HeLa细胞模型→检测过表达p53的功能与稳定性→验证E6对p53磷酸化的抑制作用→在p53/E6缺失的H1299细胞中交叉验证结论,形成完整的假设-验证闭环。
3.1 可诱导p53过表达HeLa细胞模型构建与验证
实验目的是建立剂量和时间可控的p53过表达体系,排除外源性p53表达的非特异性干扰。方法是采用DOX诱导的HTet23p53、HTet26p53细胞系(分别过表达p53)和HTet43GFP细胞系(对照),设置不同浓度DOX(0-2000ng/ml)处理48小时,或固定1000ng/ml DOX处理不同时间(1-48小时),通过蛋白质免疫印迹(Western blot)检测p53表达水平。结果显示,DOX以剂量依赖方式诱导p53过表达,2000ng/ml DOX处理后p53表达量升高5倍(n=3,P<0.05);同时以时间依赖方式诱导p53表达,48小时处理后p53表达量升高5倍(n=3,P<0.05),而对照细胞系中p53表达无显著变化。
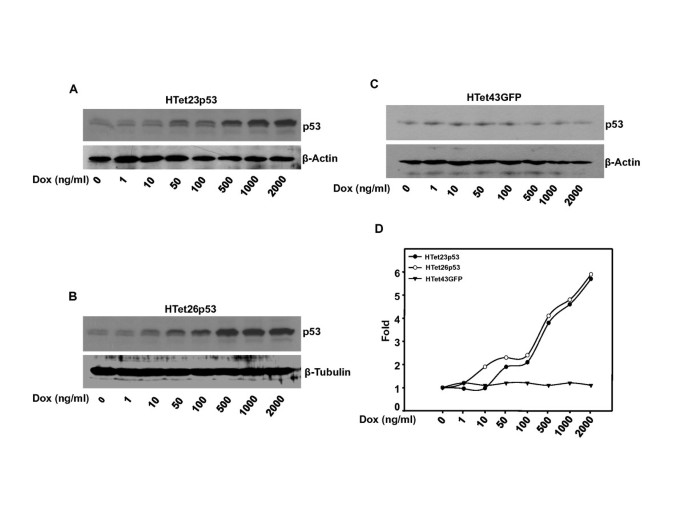
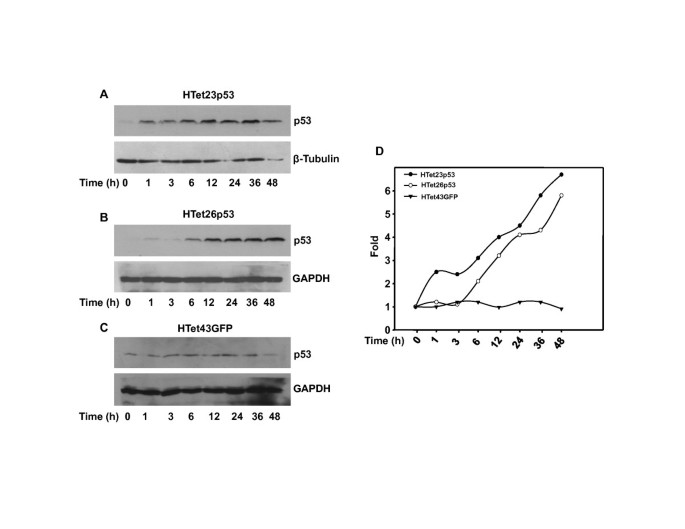
实验所用关键产品:Sigma的多西环素(Dox)、环己酰亚胺(Chx),BD的Tet系统专用血清,Santa Cruz Biotechnology的p53抗体、β-微管蛋白抗体,Cell Signaling Technology的Ser46磷酸化p53抗体。
3.2 过表达p53的肿瘤发生潜力与稳定性分析
实验目的是明确过表达p53在HPV阳性细胞中的功能状态与稳定性,排除降解途径的影响。方法包括:软琼脂糖实验检测锚定非依赖生长能力;环己酰亚胺 chase实验检测p53半衰期;蛋白酶体抑制剂MG132和乳胞素处理检测p53的蛋白酶体降解敏感性。结果显示,过表达p53的HeLa细胞锚定非依赖生长能力与对照细胞无显著差异,说明过表达p53无抑癌功能;过表达p53的半衰期为6小时,而内源性p53半衰期<1小时(n=3,P<0.01);蛋白酶体抑制剂处理可显著稳定内源性p53,但对过表达p53的水平无显著影响,提示过表达p53与内源性p53的降解调控机制存在差异。
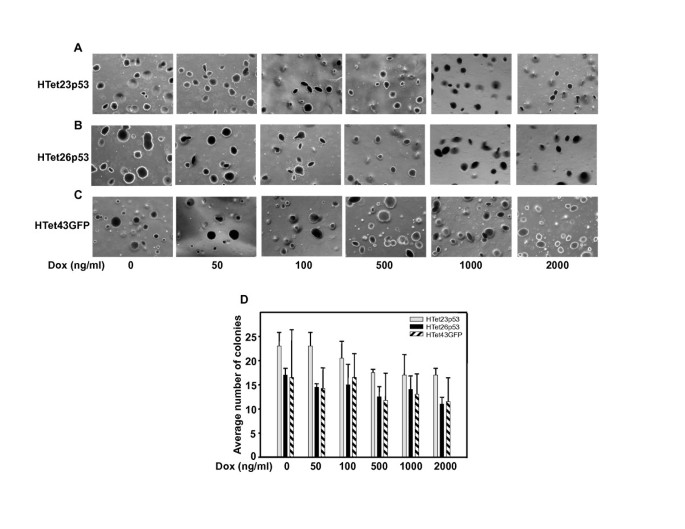
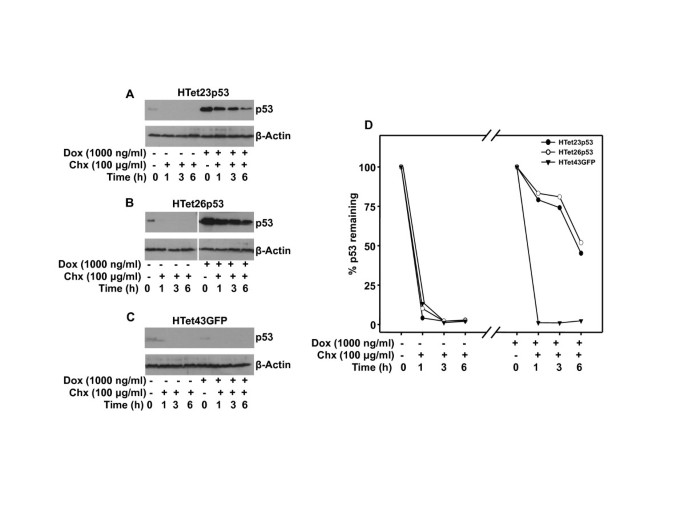
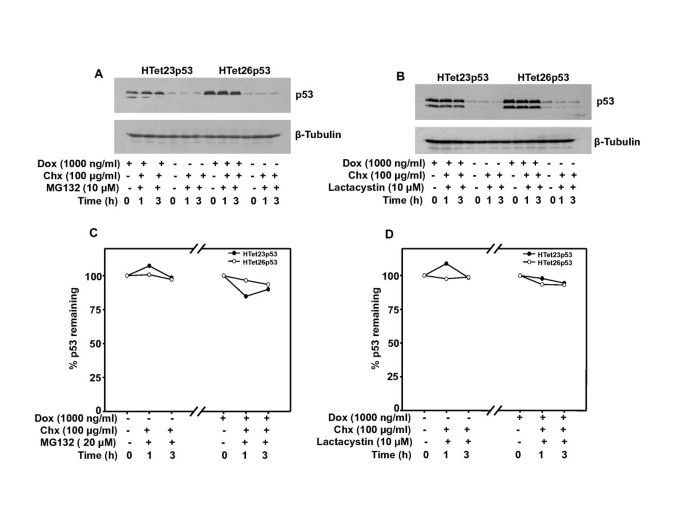
实验所用关键产品:Calbiochem的MG132、乳胞素,FMC Bioproducts的低熔点琼脂糖。
3.3 E6对p53磷酸化与功能的抑制作用验证
实验目的是验证E6通过抑制p53磷酸化失活其功能的核心假设。方法包括:用蛋白磷酸酶2A(PP2A)抑制剂冈田酸(OA)激活p53,转染HPV18 E6质粒后通过MTT实验检测细胞存活;免疫共沉淀检测E6与p53的结合;Western blot检测Ser46磷酸化p53水平;荧光素酶报告基因实验检测p21启动子活性。结果显示,OA处理可显著降低过表达p53细胞的存活率,而转染E6后细胞存活率恢复至对照水平(n=3,P<0.01);E6与过表达p53的结合水平极低,提示过表达p53以游离形式存在;E6处理不降低p53总蛋白水平,但显著降低Ser46磷酸化p53水平(n=3,P<0.05);同时E6显著抑制p21启动子的激活,说明p53的DNA结合功能被阻断。
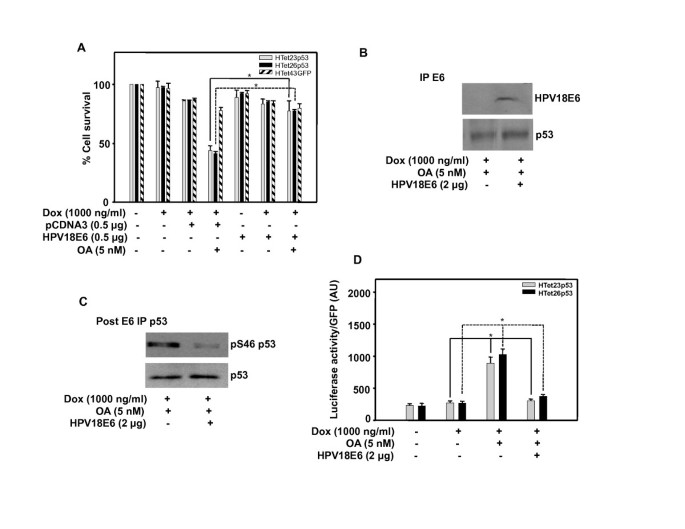
实验所用关键产品:Invitrogen的冈田酸(OA)、Lipofectamine2000转染试剂,Amersham Biosciences的荧光素酶检测试剂盒。
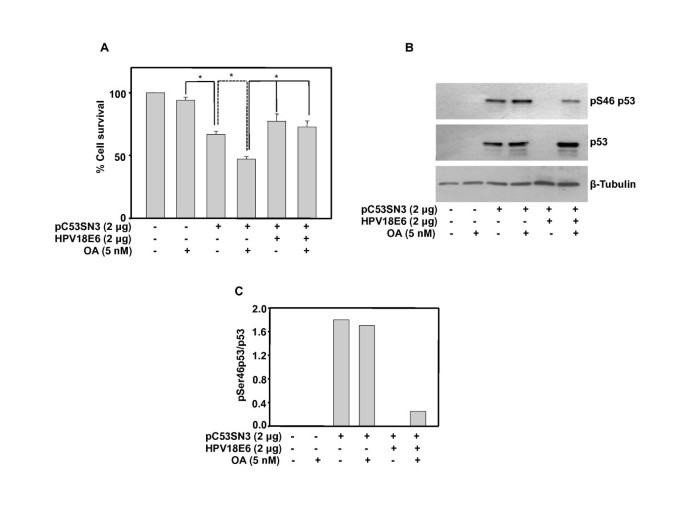
3.4 p53/E6缺失细胞系的交叉验证
实验目的是在无内源性p53和E6的细胞中验证E6对p53磷酸化的抑制作用,排除HPV阳性细胞背景的干扰。方法是在H1299细胞(p53/E6缺失)中转染p53和/或E6质粒,OA处理后通过MTT实验检测细胞存活,Western blot检测p53总蛋白与Ser46磷酸化p53水平。结果显示,过表达p53可降低H1299细胞存活率,OA处理后存活率进一步降低,而共转染E6后存活率显著恢复(n=3,P<0.01);在非OA处理细胞中E6可降解p53,但在OA处理细胞中E6不降低p53总蛋白水平,仅显著降低Ser46磷酸化p53水平,说明OA激活的p53可抵抗E6的降解,但无法抵抗E6对其磷酸化的抑制。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究中涉及的Biomarker为p53的Ser46磷酸化位点,其作为HPV阳性肿瘤中p53功能状态的关键标志物,筛选与验证逻辑为:首先在HeLa细胞中发现过表达p53功能失活与Ser46磷酸化水平降低相关,然后通过蛋白酶体抑制剂实验排除降解途径的影响,接着在H1299细胞中验证E6对该位点磷酸化的直接抑制作用,形成完整的“细胞模型发现→机制排除→交叉验证”逻辑链条。
研究过程详述
该Biomarker的来源为细胞系总蛋白样本,验证方法为Western blot检测Ser46磷酸化p53的表达水平;特异性方面,Ser46磷酸化是p53激活并发挥抑癌功能的关键修饰,仅在功能激活的p53中存在;敏感性数据显示,OA处理后Ser46磷酸化p53水平显著升高(文献未明确具体数值,基于图表趋势推测),E6处理后该水平显著降低(文献未明确具体数值,基于图表趋势推测)。
核心成果提炼
该Biomarker的功能关联为:Ser46磷酸化p53的水平直接决定p53对p21启动子的结合能力与细胞生长抑制功能,在HPV阳性细胞中,E6通过抑制该位点磷酸化导致p53功能失活;创新性在于首次在HPV相关肿瘤中发现E6非降解依赖的p53失活机制,明确了Ser46磷酸化作为p53功能状态的核心标志物;统计学结果显示,E6对细胞存活率的影响、对Ser46磷酸化p53水平的影响均具有显著统计学意义(P<0.05或P<0.01,n=3)。该成果为HPV相关肿瘤的治疗提供了新的靶点,即通过激活p53的Ser46磷酸化恢复其抑癌功能,有望突破现有靶向E6降解途径的治疗瓶颈。