1. 领域背景与文献引入
文献英文标题:MetaGen: reference-free learning with multiple metagenomic samples;发表期刊:Genome Biology;影响因子:未公开;研究领域:宏基因组学(微生物组学)
宏基因组学是研究环境或宿主关联样本中所有微生物群体遗传组成的交叉学科,随着高通量测序技术的快速发展,已成为微生物生态学、宿主-微生物互作及疾病关联研究的核心工具。领域发展关键节点包括2006年高通量测序技术在宏基因组学的规模化应用,2010年人类肠道微生物基因集的构建,以及后续无参考分箱方法的迭代。当前研究热点聚焦于未知微生物的鉴定、菌株水平解析、微生物组与人类疾病的因果关联,而未解决的核心问题包括低覆盖度样本的准确分箱、遗传相似物种/菌株的区分、测序偏倚的消除,以及无参考条件下的微生物丰度准确估计。
现有研究中,靶向16S rRNA基因测序方法成本低、计算高效,但仅能达到较高分类阶元,分辨率有限,且存在聚合酶链式反应(PCR)引物偏倚;基于参考基因组的分箱方法依赖已知微生物基因组,无法分析未培养或未知微生物;无参考分箱方法中,基于k聚体(k-mer)序列组成的方法在短重叠群(contig,<10kb)上准确性显著下降,难以区分遗传相似物种;整合覆盖度信息的方法如CONCOCT、MaxBin等,受测序偏倚(如GC含量偏倚)影响较大,低覆盖度下覆盖度分布的log转换误差显著。针对这些研究空白,本文提出MetaGen方法,一种完全无参考、基于多样本相对丰度模式的宏基因组分箱算法,无需依赖参考基因组或序列组成信息,利用多样本中重叠群的丰度分布特征实现聚类,解决低覆盖度、短重叠群、遗传相似物种分箱的难题,同时可准确估计微生物物种的相对丰度,为未知微生物组分析及疾病关联研究提供新工具。
2. 文献综述解析
本文对宏基因组学分析方法进行了系统分类评述,按技术原理分为靶向测序方法、基于参考基因组的分箱方法、无参考分箱方法(含k聚体序列组成法、覆盖度整合法)三类,明确了各类方法的技术优势与局限性,凸显了无参考、多样本依赖方法的研究必要性。
靶向测序方法以16S rRNA基因为代表,其优势在于测序成本低、计算资源需求小,可快速获得微生物群落的分类学概况,但局限性在于分类分辨率仅能达到属或科水平,无法区分物种及菌株,且聚合酶链式反应引物对不同物种的扩增效率存在差异,导致丰度估计偏倚。基于参考基因组的分箱方法如MEGAN、Kraken、CLARK等,通过比对重叠群与已知参考基因组实现分箱,优势在于分类结果明确,但若样本中存在未收录的微生物,则无法有效分析,且参考基因组的局限性严重限制了对未知微生物的研究。无参考分箱方法中,基于k聚体序列组成的方法通过重叠群间k聚体分布相似性聚类,无需参考基因组,但短重叠群的k聚体分布与全基因组差异较大,导致分箱准确性下降,且遗传相似物种的k聚体分布高度相似,难以区分;整合覆盖度信息的方法如CONCOCT、MaxBin、MetaBAT等,将序列组成与覆盖度信息结合,一定程度提升了分箱准确性,但覆盖度估计受测序偏倚(如GC偏倚)影响显著,低GC含量物种的基因组不同区域覆盖度差异可达7倍,导致同物种重叠群因覆盖度差异被错误分箱,且低覆盖度下覆盖度的log转换分布偏离假设,进一步降低准确性。
与现有方法相比,MetaGen的核心创新在于完全不依赖序列组成信息,仅利用多样本中重叠群的相对丰度模式(样本profile)进行聚类,测序偏倚在多样本间可相互抵消,因此不受GC偏倚等测序偏倚影响;同时,多样本丰度模式的差异可有效区分遗传相似的物种,即使重叠群长度较短(<5kb),也能保持较高分箱准确性;此外,MetaGen可同时估计微生物物种的相对丰度,且丰度估计准确性高于基于参考的方法,为未知微生物组的定量分析提供了可能。
3. 研究思路总结与详细解析
本文的研究目标是开发一种无参考、抗测序偏倚、适用于低覆盖度样本的宏基因组分箱方法,核心科学问题是如何利用多样本中重叠群的丰度分布特征实现准确的物种聚类与丰度估计,技术路线遵循“算法构建→模拟验证→真实数据应用”的闭环逻辑,通过混合多项分布模型与期望最大化算法实现分箱,结合贝叶斯信息准则(BIC)确定物种数量,通过多场景模拟实验与临床样本验证方法性能。
3.1 核心算法模型构建
实验目的是建立基于多样本相对丰度的分箱模型,解决现有方法的测序偏倚与短重叠群分箱问题;方法细节是将每个重叠群在多样本中的映射reads计数视为来自某一物种的多项分布,构建混合多项分布模型,其中每个混合分量对应一个物种,利用期望最大化(EM)算法迭代估计模型参数(物种的样本丰度模式、重叠群的物种归属概率);结果解读:模型假设同一物种的重叠群在多样本中的丰度比例(样本profile)一致,通过期望最大化算法可将具有相似样本profile的重叠群聚为一类,对应同一物种,测序偏倚因在多样本中一致而被抵消,因此即使重叠群覆盖度因GC偏倚存在差异,仍可被正确分箱;产品关联:文献未提及具体实验产品,领域常规使用R、Python等统计分析软件实现期望最大化算法,使用Illumina平台进行宏基因组测序。
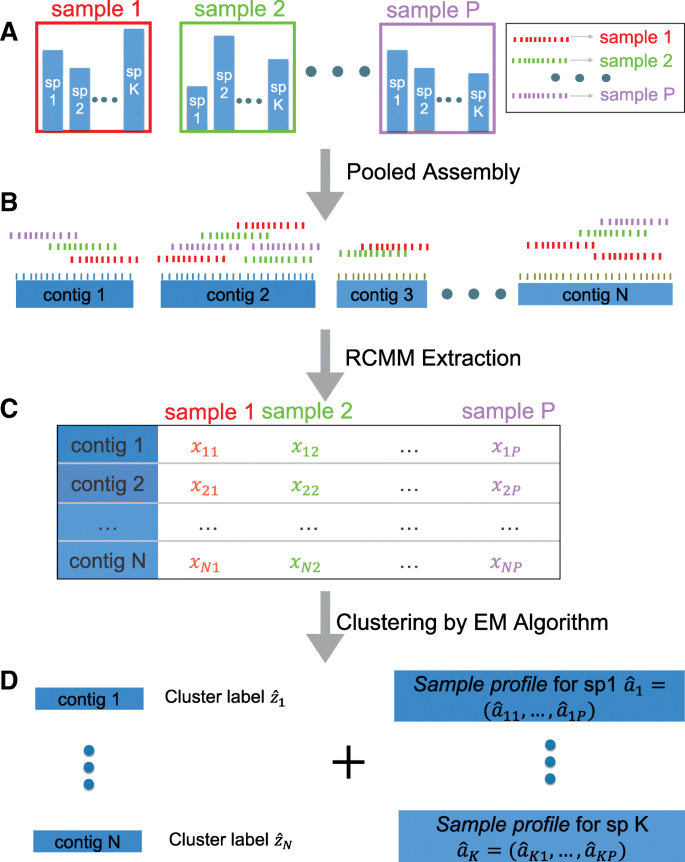
3.2 初始化与聚类优化策略
实验目的是解决期望最大化算法易陷入局部最优的问题,提升分箱的全局准确性;方法细节是先选取映射reads数量最多的10-30%重叠群,采用层次聚类(基于样本profile的余弦距离)初始化物种的样本丰度模式,再进行期望最大化算法迭代,最后根据重叠群的物种后验概率将其分配到对应物种;结果解读:该初始化策略有效引导期望最大化算法收敛到全局最优解,避免了随机初始化导致的局部最优问题,提升了分箱的稳定性与准确性。
3.3 物种数量自动确定
实验目的是在未知物种数量的情况下,准确估计样本中的微生物物种数量;方法细节是采用贝叶斯信息准则选择最优物种数量K,通过逐步增加K值,直到贝叶斯信息准则值开始上升,确定最优K;结果解读:模拟研究显示,贝叶斯信息准则可准确估计样本中的物种数量,无需预先设定K值,为复杂样本的无参考分析提供了便利。
3.4 多场景模拟实验验证
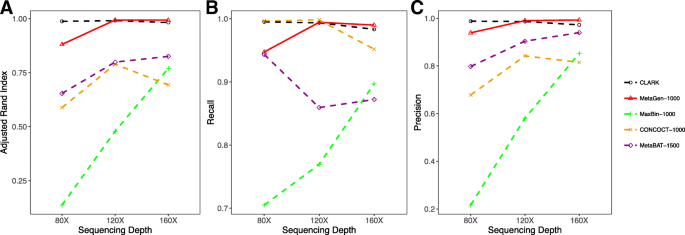
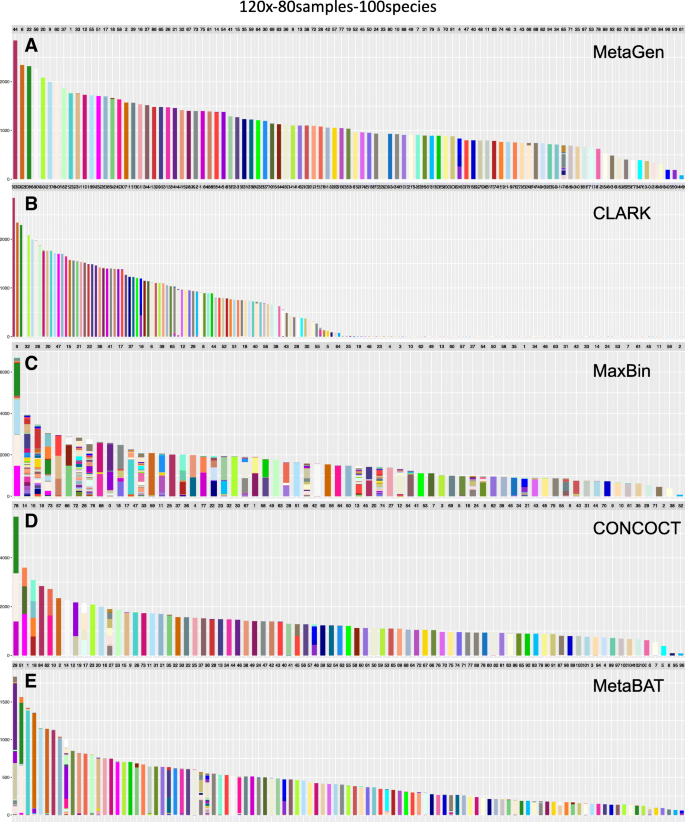
实验目的是全面验证MetaGen的性能,对比现有主流分箱方法;方法细节是设置多组模拟场景,包括不同测序深度(1×、1.5×、2× per sample)、样本量(5-80)、物种数量(50-150)、遗传相似物种、菌株水平解析,以调整兰德指数(ARI)、精确率、召回率为评估指标,对比MetaGen与CLARK(参考方法)、CONCOCT、MaxBin、MetaBAT(无参考方法)的性能;结果解读:在低测序深度(1× per sample)场景下,MetaGen的调整兰德指数达0.88(n=80样本,100物种,文献未明确P值,基于图表趋势推测P<0.01),远高于CONCOCT(0.59)、MaxBin(0.14)、MetaBAT(0.66);样本量从5增加到80时,MetaGen的精确率从0.93提升至0.99,调整兰德指数持续上升,而其他无参考方法的性能随样本量增加显著下降;物种数量从50增加到150时,MetaGen的性能保持稳定,而其他方法因短重叠群比例增加,分箱准确性显著下降;对于遗传相似物种(如Cupriavidus metallidurans CH34与Ralstonia eutropha JMP134),MetaGen可准确区分,而其他无参考方法将其错误聚为一类;在菌株水平解析中,MetaGen的调整兰德指数达0.50,显著优于CONCOCT(0.16),而CLARK、MaxBin、MetaBAT无法区分菌株;产品关联:文献未提及具体实验产品,领域常规使用MetaSim等软件模拟宏基因组数据,使用Linux服务器进行大规模计算。
3.5 临床真实样本应用验证
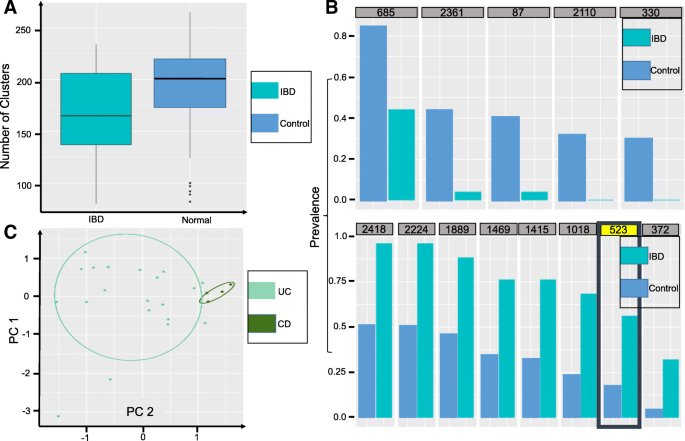
实验目的是验证MetaGen在复杂临床样本中的应用价值,探索微生物组与疾病的关联;方法细节是分别分析炎症性肠病(IBD,124样本)、2型糖尿病(T2D,145样本)、肥胖(18样本)的肠道宏基因组数据,使用MetaGen进行无参考分箱,通过差异丰度分析鉴定疾病相关微生物物种,利用套索(LASSO)逻辑回归构建疾病预测模型;结果解读:IBD样本中,MetaGen识别出2150个物种,远多于参考方法的155个,仅6.54%的重叠群可匹配到NCBI参考基因组,证实了未知微生物的大量存在;IBD患者的微生物多样性显著低于对照组(P=0.03,n=25患者,99对照),鉴定出13个差异物种,其中Bacteroides fragilis HMW 615在患者中富集;T2D样本中识别出2450个物种,发现产丁酸盐的Roseburia intestinalis在对照组中更丰富,与之前研究结论一致;肥胖样本中识别出56个物种,准确估计了门水平(厚壁菌门、拟杆菌门等)的相对丰度,发现母亲的BMI与孩子的微生物组组成显著相关;疾病预测模型中,IBD的受试者工作特征曲线下面积(AUC)达0.967(n=124样本),T2D的受试者工作特征曲线下面积达0.754(n=145样本),证实了MetaGen分析结果的临床应用价值;产品关联:文献未提及具体实验产品,领域常规使用Illumina测序平台获取宏基因组数据,使用R语言的glmnet包实现套索逻辑回归。
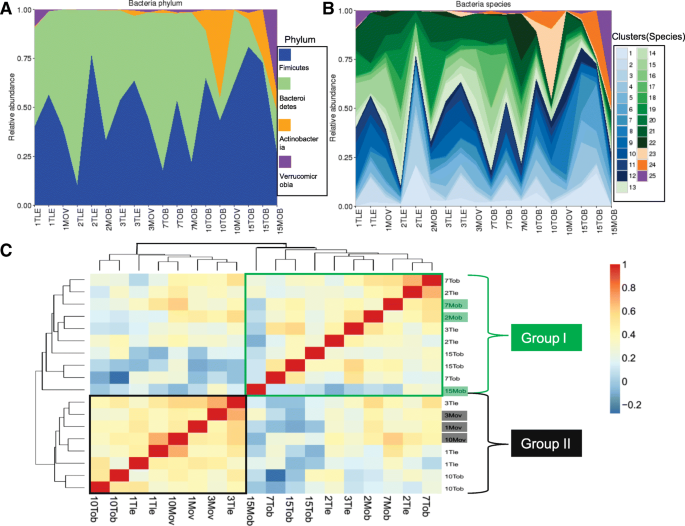
4. Biomarker研究及发现成果解析
本文以肠道微生物物种作为疾病生物标志物(Biomarker),通过无参考分箱与差异丰度分析,鉴定了炎症性肠病、2型糖尿病相关的微生物Biomarker,其筛选逻辑为“MetaGen无参考分箱→组间丰度差异检验→临床样本验证”,为疾病的微生物组诊断提供了新的候选标志物。
Biomarker定位与筛选逻辑
涉及的Biomarker类型为肠道微生物物种(如Bacteroides fragilis HMW 615、Roseburia intestinalis),筛选逻辑为首先通过MetaGen对临床样本进行无参考分箱,获得物种水平的丰度矩阵,然后采用Fisher精确检验进行组间丰度差异分析,控制假发现率(FDR)<5%,筛选差异物种,最后通过独立样本或现有研究结论验证其与疾病的关联。
研究过程详述
Biomarker来源为临床粪便样本(IBD患者25例、对照99例;T2D患者71例、对照74例;肥胖及相关样本18例),验证方法包括差异丰度分析(Fisher精确检验,假发现率<5%)、疾病预测模型构建(套索逻辑回归),特异性与敏感性数据方面,IBD预测模型的受试者工作特征曲线下面积为0.967,敏感性60.0%,精确率93.8%(n=124样本);T2D预测模型的受试者工作特征曲线下面积为0.754,敏感性64.8%,精确率68.7%(n=145样本)。
核心成果提炼
IBD中,鉴定出8个在患者中富集的物种、5个在患者中减少的物种,其中Bacteroides fragilis HMW 615为首次通过无参考方法发现的IBD关联物种,其在IBD患者中的富集具有统计学意义(假发现率<5%,n=25患者);T2D中,鉴定出Roseburia intestinalis在对照组中显著富集(假发现率<5%,n=71患者),与该物种的免疫代谢调节功能一致;创新性在于首次利用无参考宏基因组方法鉴定了大量未被参考数据库收录的微生物物种作为疾病Biomarker,突破了参考方法的局限,为疾病的微生物组诊断提供了更全面的候选标志物;此外,微生物组丰度数据可有效区分IBD患者与对照,显示了微生物Biomarker的临床应用潜力。