1. 领域背景与文献引入
文献英文标题:Saturation-scale functional evidence supports clinical variant interpretation in Lynch syndrome;发表期刊:Genome Biology;影响因子:未公开;研究领域:林奇综合征(遗传性癌症易感综合征)的基因变异解读与功能验证
林奇综合征是全球最常见的遗传性癌症易感综合征之一,每300人中约1人患病,主要关联结直肠癌和子宫内膜癌,杂合携带者的相关癌症风险分别高达80%和60%,发病时间比散发病例早数十年。该综合征由MSH2、MLH1、MSH6、PMS2四个DNA错配修复(MMR)基因的遗传缺陷导致,这些基因是临床癌症基因panel检测的核心项目。当前领域的核心瓶颈在于,临床基因检测中存在大量意义未明变异(VUS),尤其是错义变异,约三分之一的林奇综合征基因变异仍为VUS,且传统重分类率仅约25%,这严重限制了检测结果的临床应用价值,无法为患者和家属提供明确的风险评估与诊疗指导。
现有研究中,传统变异分类方法依赖人群频率、肿瘤特征、共分离分析等正交证据,但对罕见错义VUS的分类能力有限;多重变异效应分析(MAVE)作为高通量功能检测技术,已在BRCA1、TP53等癌症易感基因中显示出较高的变异分类准确性,但此前在林奇综合征中的临床应用案例较少,且缺乏大规模临床队列的独立验证,尤其是针对MSH2基因的饱和功能分析尚未系统应用于VUS分类。针对这一研究空白,本研究利用覆盖MSH2基因94%以上错义变异的MAVE功能图谱,结合15520例林奇综合征基因变异患者的临床数据库,开展大规模变异重分类研究,旨在验证MAVE功能数据的临床有效性,解决VUS分类瓶颈,为临床诊疗和风险评估提供精准依据。
2. 文献综述解析
本研究的文献综述按研究技术类型(传统变异分类方法、高通量MAVE技术)和研究应用场景(不同癌症易感基因的VUS分类)对现有研究进行分类,系统梳理了林奇综合征变异分类的现状、MAVE技术的优势与局限性,明确了当前领域的核心研究空白。
传统变异分类方法基于人群频率、肿瘤特征、共分离分析等正交证据,能对部分明确的致病性或良性变异进行分类,但对于罕见错义VUS的分类能力不足,导致大量变异无法得到明确临床解读;MAVE技术可高通量检测变异的功能效应,已在部分癌症易感基因中展现出高效的VUS分类潜力,但此前在林奇综合征中的应用缺乏大规模临床队列验证,且部分研究存在循环验证风险(即使用依赖功能证据的变异作为对照),同时尚未充分利用肿瘤-正常配对样本数据解析体细胞变异模式。
本研究的创新价值在于,首次在大规模林奇综合征临床队列中验证了MSH2基因MAVE功能图谱的临床有效性,且在验证过程中排除了依赖功能证据分类的变异,彻底避免了循环验证的风险;同时结合MAVE功能数据与SpliceAI剪接预测数据,实现了682个MSH2基因错义VUS的大规模重分类,显著提升了VUS分类效率;此外,本研究还将MAVE功能数据拓展应用于肿瘤-正常配对样本分析,识别出散发性林奇样综合征患者,为这类患者的诊疗和家属风险评估提供了新的技术路径。
3. 研究思路总结与详细解析
本研究的核心目标是验证MSH2基因MAVE功能图谱在林奇综合征变异分类中的临床有效性,解决VUS分类瓶颈;核心科学问题是MAVE功能数据能否作为“强”功能证据用于MSH2基因变异分类,以及如何结合临床数据实现大规模VUS重分类;技术路线遵循“功能图谱构建→临床数据库整合→有效性验证→VUS重分类→临床关联分析→体细胞变异解析”的闭环逻辑。
3.1 MSH2功能图谱与临床数据库整合
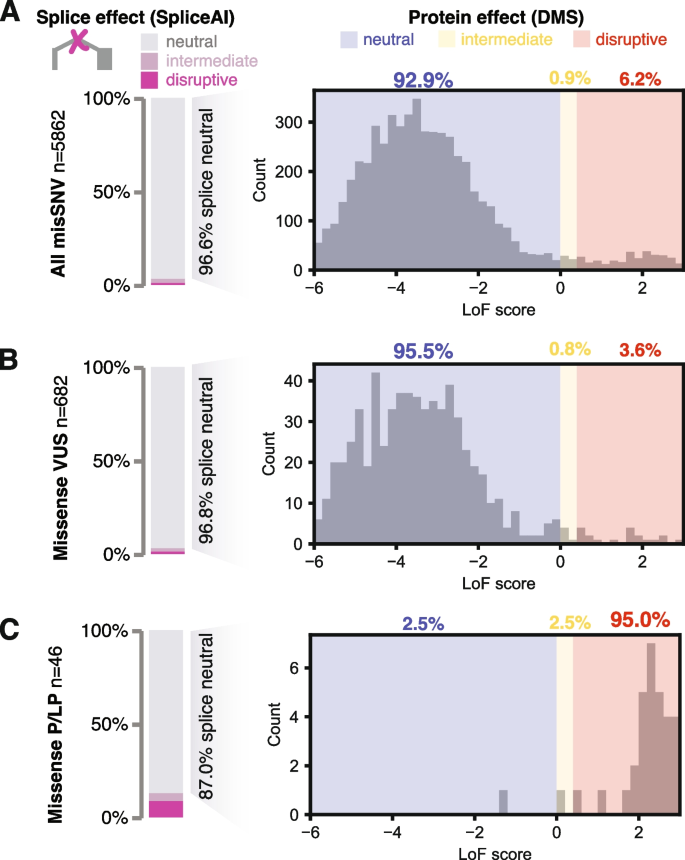
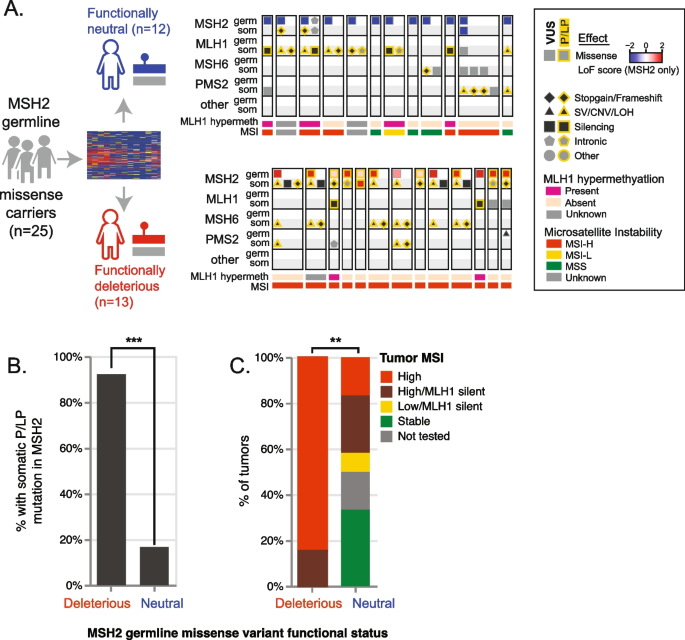
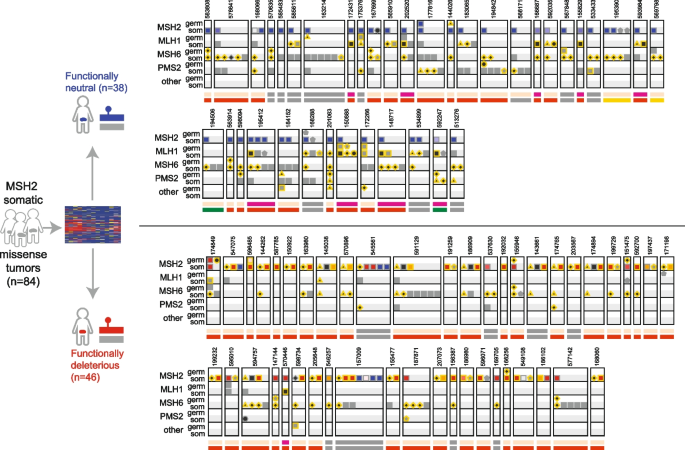
实验目的是将MSH2基因的MAVE功能图谱与林奇综合征患者临床数据库整合,为后续变异验证和分类提供数据基础。研究采用覆盖MSH2基因16749个错义变异的MAVE功能图谱(来自此前的深度突变扫描研究),整合包含15520例林奇综合征基因变异患者的临床数据库,其中1604例为肿瘤-正常配对样本,13916例为仅生殖系检测样本;对每个MSH2错义变异,同时注释MAVE的功能丧失(LoF)评分和SpliceAI的剪接破坏评分,定义LoF评分≥0.4或SpliceAI评分≥0.5为功能异常,LoF评分<0且SpliceAI评分<0.2为功能正常,中间值为中间型。结果显示,整合后的数据覆盖了大量MSH2基因变异,其中94.4%的MSH2错义变异携带者携带的是VUS(1829例患者,682个独特VUS)。文献未提及具体实验产品,领域常规使用的MAVE技术相关试剂包括基因合成试剂盒、高通量测序平台等,SpliceAI为公开的生物信息学工具。
3.2 MSH2功能图谱的临床验证
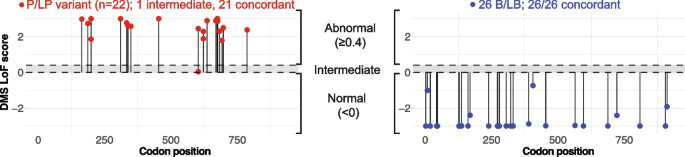
实验目的是验证MSH2基因MAVE功能数据的临床有效性,确定其作为变异分类证据的强度。研究从临床数据库中筛选出47个已通过正交证据(不依赖功能数据)分类的MSH2错义变异作为对照,其中22个为致病性/可能致病性,25个为良性/可能良性;计算MAVE功能数据与临床分类的一致性,通过OddsPath评分量化功能证据的强度,并验证其在独立验证集中的召回率。结果显示,MAVE功能测量结果与47个对照变异的临床分类完全一致(除1个致病性变异处于中间范围),OddsPath评分显示功能异常评分的证据强度为24.9,功能正常评分的证据强度为0.043,达到了ACMG/AMP指南中的“强”功能证据标准(PS3/BS3);在独立验证集中,MSH2错义功能评分对致病性变异的召回率为96.9%。
3.3 MSH2错义VUS的重分类
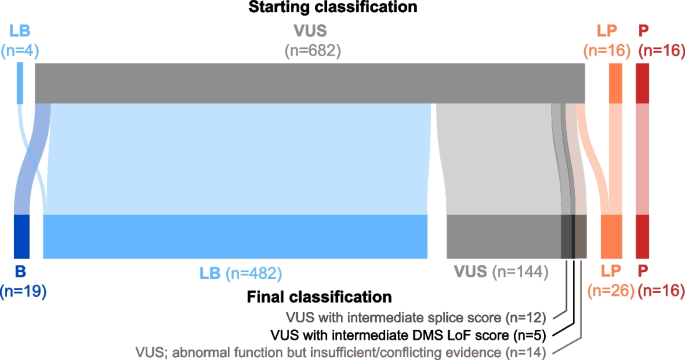
实验目的是利用MAVE功能数据对MSH2基因错义VUS进行重分类,解决VUS分类瓶颈。研究对682个独特的MSH2错义VUS,根据MAVE LoF评分和SpliceAI评分进行功能分类,结合ACMG/AMP指南添加功能证据代码,评估变异重分类的可能性;对于功能异常的VUS,进一步结合临床证据(如肿瘤MMR缺陷检测结果)进行正式重分类;对于功能正常的VUS,评估添加BS3证据代码后能否重分类为良性/可能良性。结果显示,682个VUS中,5.0%(34个)功能异常,其中10个被正式重分类为致病性/可能致病性,497个功能正常的VUS可在添加BS3证据后重分类为良性/可能良性,约74%的VUS可实现新分类。
3.4 功能异常变异与癌症风险的关联分析
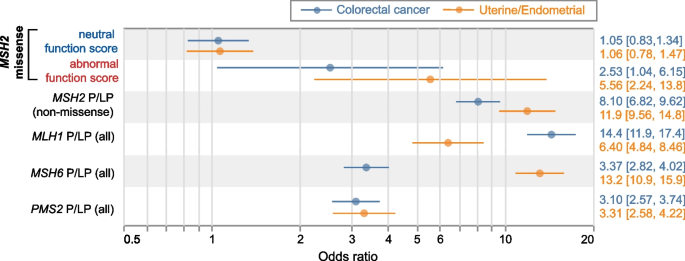
实验目的是分析MSH2基因功能异常错义变异与林奇综合征相关癌症风险的关联,验证功能数据的临床相关性。研究将MSH2错义变异携带者按功能状态分为功能异常组和功能正常组,采用逻辑回归模型分析两组患者的结直肠癌和子宫内膜癌风险,与已知的致病性/可能致病性变异组进行比较。结果显示,功能异常的MSH2错义变异携带者的结直肠癌风险显著升高(OR=2.53,95%CI:[1.04,6.15],P=0.04),子宫内膜癌风险也显著升高(OR=5.56,95%CI:[2.24,13.8],P=2.2×10^-4),但风险低于截短型致病性/可能致病性变异;功能正常的变异携带者未显示出显著的相关癌症风险。
3.5 肿瘤-正常配对样本的变异模式分析
实验目的是应用MAVE功能数据解析肿瘤-正常配对样本中的生殖系和体细胞变异模式,识别散发性林奇样综合征患者。研究在1604例肿瘤-正常配对样本中,筛选出仅携带MSH2错义生殖系变异的患者,按生殖系变异的功能状态分为功能异常组和功能正常组,分析两组患者的体细胞“二次打击”发生率、肿瘤微卫星不稳定性(MSI)状态,以及体细胞变异与其他MMR基因的关联。结果显示,携带功能异常MSH2生殖系变异的患者中,92.4%存在致病性/可能致病性的体细胞“二次打击”,而功能正常组仅16.7%存在该情况(P=0.00021);功能异常组的肿瘤均为MSI高,而功能正常组仅部分为MSI高;此外,识别出29例携带双等位基因体细胞功能丧失变异的患者,属于散发性林奇样综合征,其家属无需进行额外的癌症筛查。
4. Biomarker研究及发现成果解析
本研究中的Biomarker为MSH2基因的功能异常错义变异,其筛选逻辑为:基于MAVE深度突变扫描的LoF评分和SpliceAI剪接预测评分筛选功能异常变异,再结合临床队列的肿瘤特征、癌症风险数据及肿瘤-正常配对样本的体细胞变异模式进行多维度验证,最终确定其作为林奇综合征风险评估和变异分类的功能Biomarker。
该Biomarker的来源为MSH2基因的错义变异,来自15520例林奇综合征基因变异患者的临床检测数据;验证方法包括临床队列的变异分类一致性验证、癌症风险的逻辑回归分析、肿瘤-正常配对样本的体细胞变异分析及肿瘤MSI状态检测;特异性与敏感性数据显示,MAVE功能数据对已知致病性/可能致病性变异的召回率为96.9%,功能异常变异与结直肠癌风险的关联OR值为2.53(95%CI:[1.04,6.15],P=0.04),与子宫内膜癌风险的关联OR值为5.56(95%CI:[2.24,13.8],P=2.2×10^-4),与体细胞“二次打击”的关联具有显著统计学差异(P=0.00021)。
核心成果方面,该Biomarker明确为林奇综合征的致病性变异,携带者的结直肠癌和子宫内膜癌风险显著升高,且肿瘤易呈现MSI高状态;其创新性在于首次在大规模临床队列中建立了MSH2基因饱和功能变异与临床表型的关联,实现了VUS的高效重分类,同时识别出散发性林奇样综合征的体细胞变异模式,为临床精准诊疗和家属风险分层提供了关键依据。