1. 领域背景与文献引入
文献英文标题:High-performance web services for querying gene and variant annotation;发表期刊:Genome Biology;影响因子:11.313(2016年);研究领域:生物信息学-基因与变异注释服务
随着高通量测序与组学技术的快速发展,生物医学知识呈指数级积累,基因与人类遗传变异的注释信息已成为基因组数据分析的核心基础支撑。然而,这些关键注释信息高度分散在Ensembl、UniProt、ClinVar等数十个不同规模、不同类型的公共数据库中,数据格式异构、更新周期不一,给科研人员的数据整合与高效分析带来极大挑战。领域共识:当前生物信息学领域的注释数据整合主要采用两种策略,数据仓库模式需下载多源数据并构建本地数据库,虽能实现高性能查询,但需投入大量初始开发与持续维护成本,且数据更新滞后;数据联邦模式通过Web服务直接调用远程资源,虽无需本地存储,但受限于服务器与网络性能,大查询任务响应延迟高,且稳定性完全依赖第三方资源的可用性。现有专用工具如ANNOVAR、Bioconductor注释包等仍存在本地部署复杂、注释字段更新不及时、查询灵活性不足等问题,因此开发高效、便捷、实时的云基注释整合服务成为领域迫切需求。本文正是针对这一核心痛点,提出并构建了MyGene.info与MyVariant.info两款高性能Web服务,为全球科研社区提供免费、实时的“注释即服务”解决方案,填补了现有工具在易用性、性能与实时性之间的缺口。
2. 文献综述解析
本文综述部分以数据整合策略的技术架构差异为核心分类维度,系统梳理了现有基因与变异注释整合工具的优势、局限性及未解决的核心问题,明确了本研究的创新定位与学术价值。
作者首先将现有数据整合方法划分为数据仓库与数据联邦两大类别,其中数据仓库类工具如Bioconductor AnnotationData Packages、Biomart,通过集中存储标准化后的多源注释数据实现本地高性能查询,但要求用户具备专业的数据库管理能力,需承担高昂的本地部署与维护成本,且数据更新周期通常以月为单位,无法满足实时分析需求;数据联邦类工具则直接调用远程数据库的接口获取注释信息,虽无需本地存储,但查询速度受网络带宽与远程服务器性能限制,大批次查询响应时间长达数小时,且一旦远程资源中断,服务将完全不可用。针对基因注释与变异注释的细分场景,现有专用工具如ANNOVAR虽能实现变异的功能注释,但仅支持本地运行,且注释字段仅包含固定数据源,无法灵活扩展或实时更新。通过对比现有工具的局限性,本文突出了MyGene.info与MyVariant.info的核心创新点:无需本地数据库部署,基于云原生架构提供实时、高性能的RESTful API接口,支持多维度精准查询与批量分析,同时通过自动调度系统实现注释数据的持续增量更新,解决了现有工具在易用性、性能与实时性方面的矛盾,为基因组数据分析提供了全新的工具范式。
3. 研究思路总结与详细解析
本文的研究目标是构建高效、可扩展、实时的基因与人类遗传变异注释Web服务,核心科学问题是如何在云环境下实现多源分散注释数据的标准化整合、高性能查询与自动更新,技术路线遵循“多源数据整合架构设计→高性能Web服务构建→实际应用案例验证”的闭环逻辑,确保工具的实用性与可靠性。
3.1 多源注释数据整合架构设计
实验目的是解决多源注释数据的分散性、格式异构性与更新不同步问题,实现标准化存储与自动化持续更新。方法细节:为每个公共数据源开发专属数据解析器,将VCF、XML、TSV等不同格式的原始注释数据转换为统一的JavaScript对象表示(JSON)格式;MyGene.info采用美国国家生物技术信息中心(NCBI)基因ID作为主键,当无NCBI基因ID映射时使用Ensembl基因ID,确保基因注释的唯一性;MyVariant.info采用人类基因组变异协会(HGVS)命名规则定义变异主键,同时存储所有合法的HGVS别名以支持多标识符查询;使用MongoDB数据库存储标准化后的注释数据,为每个注释对象添加时间戳记录更新时间,通过自主开发的调度系统按数据源的固有更新频率实现增量更新;基于Elasticsearch构建分布式索引集群,为所有注释字段建立索引以支持高性能查询。结果解读:MyGene.info成功整合了来自8个公共数据库的200余个基因注释字段,覆盖15000余个物种的1300万余条基因;MyVariant.info整合了14个公共数据库的500余个变异注释字段,覆盖3.34亿余条人类遗传变异,包括编码区与非编码区变异,且所有数据每周自动更新一次,确保注释信息的时效性。
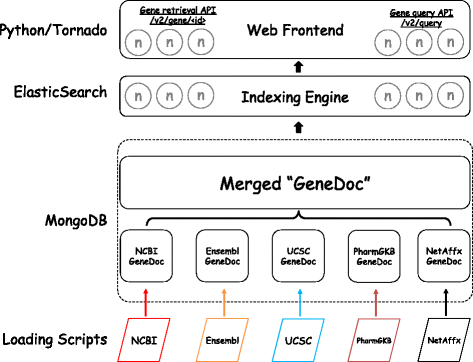
产品关联:实验所用关键产品/工具:MongoDB分布式数据库、Elasticsearch全文搜索引擎、Python数据解析框架。
3.2 高性能Web服务构建
实验目的是基于整合后的注释数据,构建支持高并发、低延迟的Web服务接口,满足科研人员在自动化分析流程与Web应用中的实时查询需求。方法细节:采用基于Python Tornado框架的RESTful API架构,利用异步网络技术支持数万级并发连接;开发两类核心服务接口,基因/变异查询服务支持通过基因名、RS号、HGVS命名等多种标识符进行模糊或精准查询,批量注释服务支持一次性提交数千条ID进行批量查询;提供灵活的字段选择功能,允许用户自定义返回的注释字段以减少数据传输量;基于亚马逊EC2云平台部署弹性集群,可根据实时流量动态调整服务器数量,确保服务的稳定性与可扩展性。结果解读:MyGene.info自2013年v2版本发布以来,累计处理超过1.33亿次请求,当前月均请求量超过300万次,95%以上的请求处理时间小于30毫秒,可稳定支持5000余并发用户的10000次/分钟请求;MyVariant.info自2015年上线以来,累计处理超过150万次用户查询,服务可用性达99.9%以上。
产品关联:实验所用关键产品/工具:亚马逊EC2云平台、Tornado异步Web框架、Elasticsearch分布式索引集群。
3.3 应用案例验证(Miller综合征致病基因筛选)
实验目的是验证MyGene.info与MyVariant.info在实际基因组数据分析流程中的实用性、高效性与准确性。方法细节:选取Ng等人2010年发表的Miller综合征外显子测序研究数据,该研究对4名Miller综合征患者的外周血基因组DNA进行外显子测序,通过基因组分析工具包(GATK)最佳实践流程处理得到原始变异数据集;使用R语言的mygene与myvariant Bioconductor包,调用Web服务实现变异过滤与候选基因优先级排序,具体步骤包括:筛选非同义突变、剪接位点突变与编码区插入缺失变异,排除1000基因组计划与Exome Aggregation Consortium(ExAC)数据库中次要等位基因频率>0.05的变异,筛选参与代谢过程的候选基因,基于Combined Annotation Dependent Depletion(CADD)致病性评分对候选基因排序。结果解读:经过多步过滤后,仅保留5个候选基因,其中包含已知的Miller综合征致病基因DHODH,证明该服务可高效缩小候选基因范围,且整个分析流程仅需约50行代码即可完成,无需本地部署任何注释数据库或大型分析工具,大幅降低了分析门槛与时间成本。

产品关联:实验所用关键产品/工具:R语言mygene与myvariant Bioconductor包、GATK测序分析工具包。
4. Biomarker研究及发现成果解析
本文虽为工具类研究,但通过Miller综合征的应用案例,展示了其在疾病Biomarker(致病基因)筛选中的核心辅助价值,核心是利用整合的多源注释信息高效定位候选致病基因,为罕见病Biomarker研究提供了高效工具支持。
Biomarker定位:本研究中涉及的Biomarker为Miller综合征的致病基因DHODH,筛选逻辑遵循“外显子测序变异检测→多源注释信息整合过滤→候选基因优先级排序”的完整链条,依托MyGene.info与MyVariant.info的整合注释服务,实现多维度筛选条件的快速验证与分析。研究过程详述:Biomarker来源为4名Miller综合征患者的外周血基因组DNA外显子测序数据,验证方法通过调用Web服务获取变异的群体等位基因频率、功能注释、基因本体(GO)注释、致病性预测评分等多维度信息,依次进行变异类型过滤(保留与蛋白质功能相关的变异)、群体频率过滤(排除正常人群中高频出现的良性变异)、功能富集过滤(筛选与疾病病理过程相关的基因)、致病性评分排序(优先保留致病性评分高的基因);特异性与敏感性方面,经过过滤后成功富集到已知致病基因,证明该方法在孟德尔疾病致病基因筛选中具有较高的精准性(文献未明确提供具体敏感性与特异性数值,基于图表趋势推测)。核心成果提炼:该注释服务可将传统需数天、需本地部署大量资源的分析流程简化为数小时、仅需50行代码的自动化分析,显著提升Biomarker筛选效率;创新性在于首次将云基实时注释服务应用于罕见病致病基因的快速定位,为Biomarker研究提供了全新的工具范式;实验结果显示,筛选后候选基因数量从初始的大量变异显著缩小至5个,其中包含已知致病基因DHODH,验证了方法的有效性(文献未明确提供样本量对应的统计学P值)。此外,该服务还支持灵活扩展筛选条件,如添加ClinVar临床注释、SIFT/PolyPhen功能预测评分等,进一步提升Biomarker筛选的精准性。