1. 领域背景与文献引入
文献英文标题:Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads;发表期刊:Genome Biology;影响因子:未公开;研究领域:转录组学、功能基因组学。
RNA测序技术在2000年代末成为转录组分析的核心工具,相比传统微阵列技术具有更高的动态范围,能直接检测可变剪接产生的转录本异构体,还可通过序列信息分析等位基因表达差异。然而,早期RNA测序数据分析方法存在两大核心局限:一是无法同时估计转录本异构体表达水平与等位基因表达不平衡,现有方法仅能单独处理这两类生物学问题;二是多映射reads(可比对到多个转录本或基因的reads)的处理方式存在缺陷,丢弃这类reads会导致大量信息丢失,而部分分配方法无法准确估计转录本异构体的表达水平。在二倍体生物中,单倍型-转录本异构体水平的表达分析对解析顺式调控与反式调控的差异、基因组印记等表观遗传效应至关重要,但这一领域的研究方法仍处于空白阶段。
针对上述研究空白,本研究开发了一套全新的RNA测序数据分析流程与统计模型MMSEQ,旨在实现同时估计单倍型与转录本异构体的表达水平,解决多映射reads的信息利用问题,为功能基因组学研究提供更精准的分析工具。
2. 文献综述解析
作者将现有研究分为转录本异构体表达估计方法、等位基因表达不平衡分析方法两大类,系统梳理了两类方法的技术优势与局限性,明确了当前研究的核心空白。
在转录本异构体表达估计领域,现有方法可分为基于区域计数的模型与基于单reads的模型两类。基于区域计数的方法如POEM,仅利用外显子区域的reads,无法处理跨外显子或多基因的多映射reads;Jiang等的方法扩展了区域定义,纳入外显子连接区域,但同样不处理多基因的多映射reads。基于单reads的方法如RSEM,能处理多基因与多转录本的reads,但采用Bootstrap估计标准误导致计算量极大,且不兼容双端测序数据;Cufflinks虽兼容双端数据,能进行转录本组装与表达估计,但未考虑序列特异性的read生成偏差,且对多映射reads采用固定加权策略,无法准确估计转录本表达水平。
在等位基因表达不平衡分析领域,现有方法多聚焦于单个SNP位点的二项式检验,仅能验证单个位点的等位基因表达差异;部分扩展方法如针对多个SNP的检验,虽能整合同一基因内的多个SNP信息,但仍停留在基因水平的分析,无法区分同一基因不同转录本异构体的等位基因表达差异,也未与转录本异构体的表达估计相结合。
通过对比现有研究的局限性,本研究的核心创新点在于首次开发了能同时估计单倍型与转录本异构体表达水平的统计模型MMSEQ,该模型充分利用多映射reads的信息,兼容双端测序数据,能校正序列特异性的read生成偏差,还能在转录本异构体水平解析等位基因表达不平衡,填补了该领域的研究空白。
3. 研究思路总结与详细解析
本研究的核心目标是开发一套完整的RNA测序数据分析流程与统计模型,实现单倍型与转录本异构体水平的表达估计;核心科学问题是如何有效利用多映射reads,同时解析转录本异构体表达与等位基因表达不平衡,解决现有方法的技术局限;技术路线采用“流程构建-模型开发-多维度验证”的闭环逻辑,先构建个性化转录组参考的分析流程,再开发MMSEQ统计模型,最后通过模拟数据、人类HapMap数据、小鼠杂交数据验证方法的准确性与实用性。
3.1 个性化转录组分析流程构建
实验目的是建立从原始RNA测序数据到单倍型-转录本异构体表达估计的完整分析流程,解决传统参考基因组比对中因等位基因差异导致的reads丢失问题;方法细节:流程包含两种应用模式,一是基于预定义转录组参考的直接分析,二是基于数据构建个性化单倍型转录组参考的分析,后者的步骤为:使用TopHat将reads比对到标准基因组参考,通过SAMtools pileup进行基因型识别,利用polyHap结合群体基因型数据进行单倍型分型,编辑标准转录组参考以匹配个体单倍型,最后使用Bowtie将reads重新比对到个性化单倍型转录组参考;结果解读:在人类HapMap数据集中,重新比对后恢复了约0.3%的原本无法比对的reads,部分转录本获得了高达13%的额外比对结果,有效提升了数据利用率;产品关联:文献未提及具体实验产品,领域常规使用TopHat、SAMtools、polyHap、Bowtie等生物信息学工具。
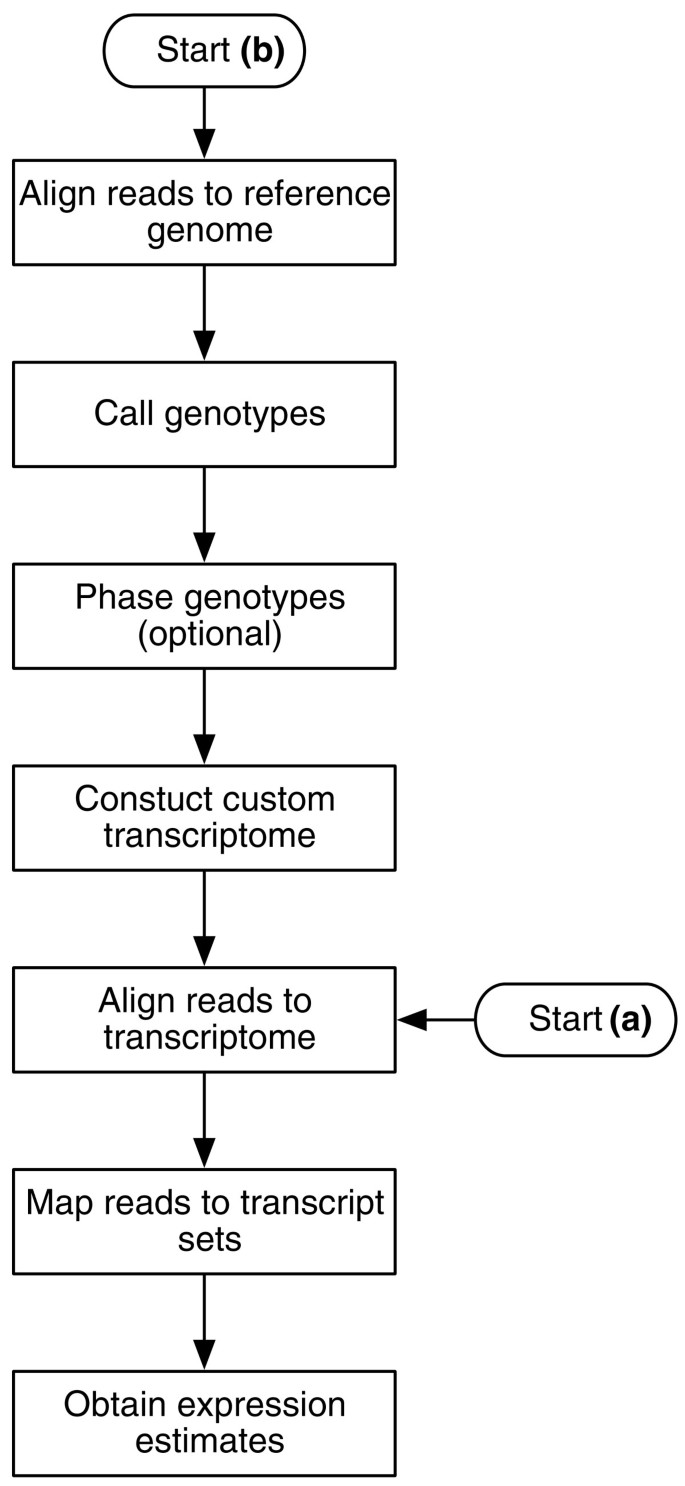
3.2 MMSEQ统计模型开发与优化
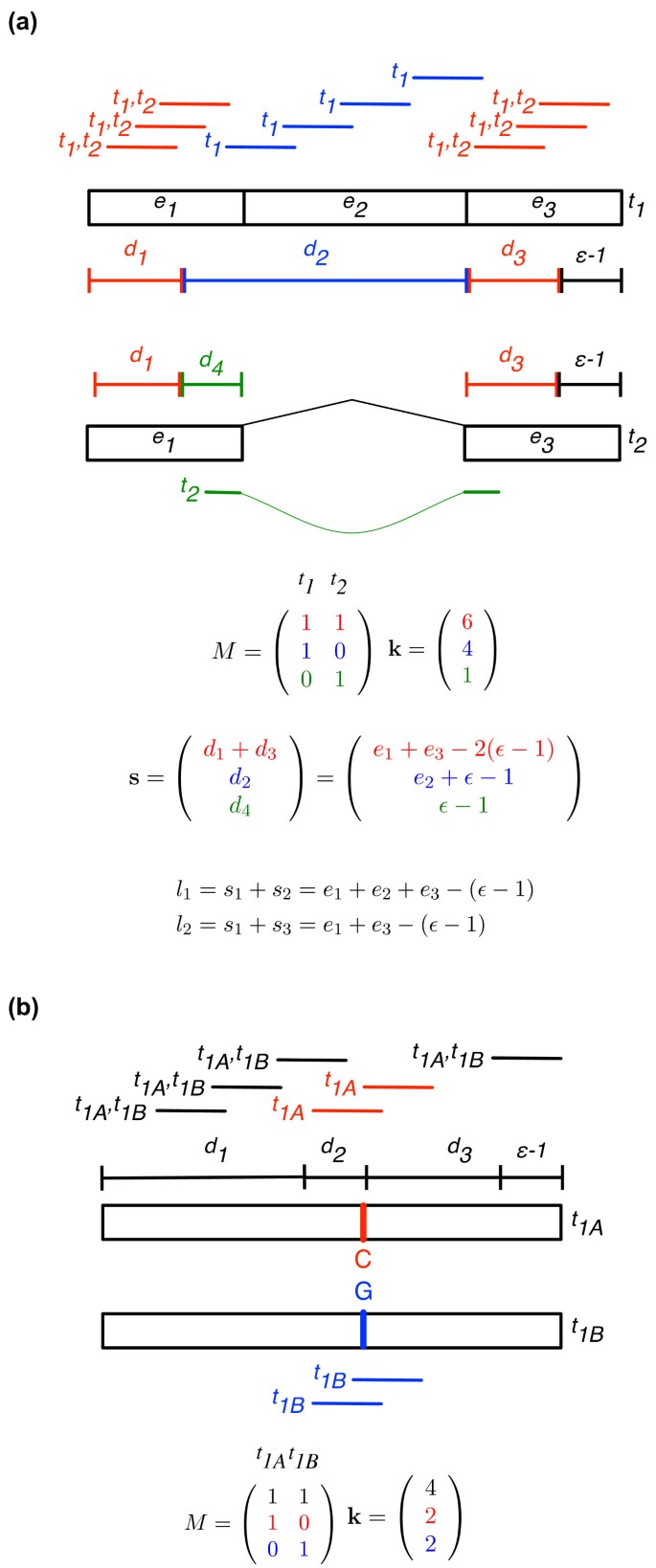
实验目的是开发能处理多映射reads、同时估计单倍型与转录本异构体表达的统计模型,校正read生成的非均匀性偏差;方法细节:模型基于泊松分布构建,将reads映射的转录本集合定义为“区域”,扩展到单倍型-转录本异构体水平;采用期望最大化(EM)算法求解最大似然估计,同时引入贝叶斯Gibbs抽样模型进行正则化估计,以降低低表达转录本的估计误差;加入最佳错配层过滤以确保reads正确分配到对应单倍型,针对双端数据加入插入片段长度过滤以提升比对准确性;使用Li等的泊松回归模型校正序列特异性的read生成偏好;结果解读:Gibbs抽样的正则化估计相比EM估计误差更低,能避免低表达转录本的低估,仅对极低表达转录本存在轻微高估;序列偏好校正后,部分转录本的表达估计发生显著变化,说明该校正步骤的必要性;产品关联:文献未提及具体实验产品,领域常规使用R、Python等统计编程环境实现算法。
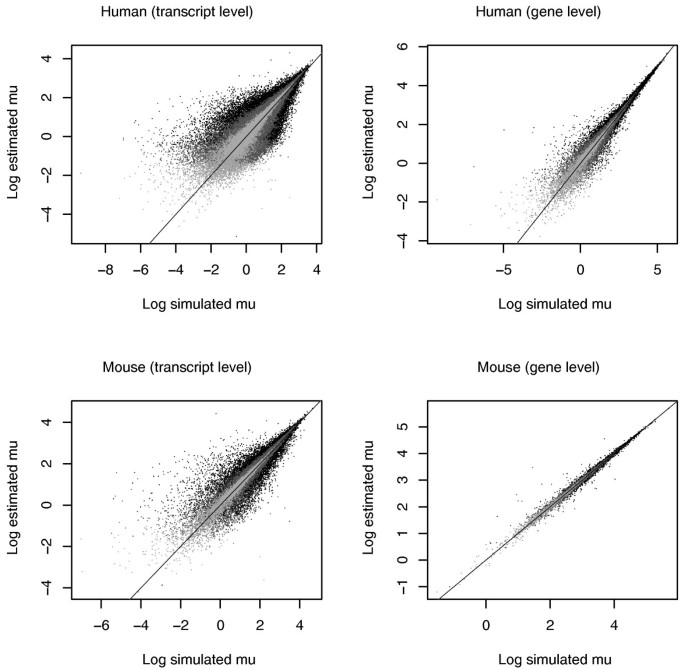
3.3 模拟数据验证方法性能
实验目的是验证MMSEQ在转录本异构体表达估计上的准确性与稳定性;方法细节:基于人类和小鼠Ensembl cDNA序列模拟测序reads,分别使用MMSEQ与RSEM进行表达估计,比较模拟值与估计值的相关性;结果解读:MMSEQ的Gibbs估计与模拟值的相关性更高,对中低表达转录本的估计更准确;RSEM与MMSEQ EM算法均存在低估低表达转录本的问题,而Gibbs估计通过正则化有效缓解了这一偏差;小鼠转录组的估计准确性高于人类,原因是人类转录组的可变剪接复杂度更高;产品关联:文献未提及具体实验产品,领域常规使用模拟测序数据的工具(如Flux Simulator)。
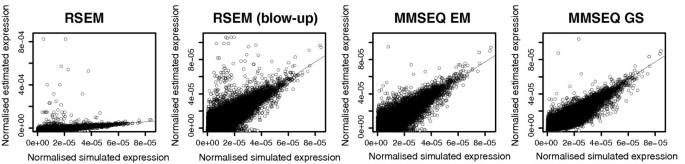
3.4 人类HapMap数据集验证重复性
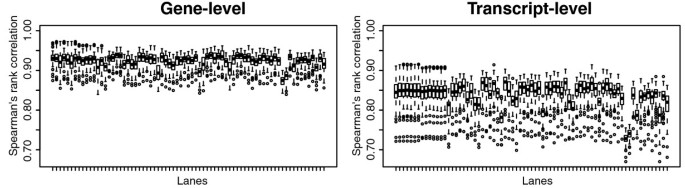
实验目的是验证MMSEQ在真实人类样本中的技术重复性与生物重复性;方法细节:分析HapMap项目的73个RNA测序样本,包括技术重复与生物重复,计算基因与转录本异构体水平的表达相关性;结果解读:基因水平的平均Spearman秩相关为0.92,转录本异构体水平为0.84;技术重复的相关性更高(基因水平0.92-0.97,转录本水平0.90-0.92),生物重复的变异符合负二项分布,说明方法的技术重复性良好,能有效区分技术变异与生物变异;产品关联:文献未提及具体实验产品,领域常规使用HapMap公共数据库的测序数据。
3.5 单倍型-转录本异构体解析能力验证
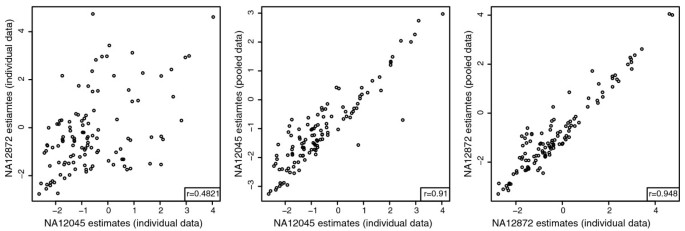
实验目的是验证MMSEQ能准确解析同一基因两个单倍型的转录本异构体表达差异;方法细节:选取人类HapMap数据中两名男性样本的X染色体非拟常染色体区域(单倍体),构建包含两个单倍型的混合转录组参考,模拟女性样本的两个X染色体,比较单独样本的表达估计与混合样本的解析结果;人为制造90%的等位基因表达不平衡,验证方法在极端不平衡下的解析能力;结果解读:即使在人为制造的极端表达不平衡下,混合样本的解析结果与单独样本的估计值相关性仍达0.91-0.95,说明MMSEQ能准确解析单倍型特异性的转录本异构体表达;产品关联:文献未提及具体实验产品,领域常规使用人类基因组公共数据库数据。
3.6 小鼠杂交数据集验证生物学应用
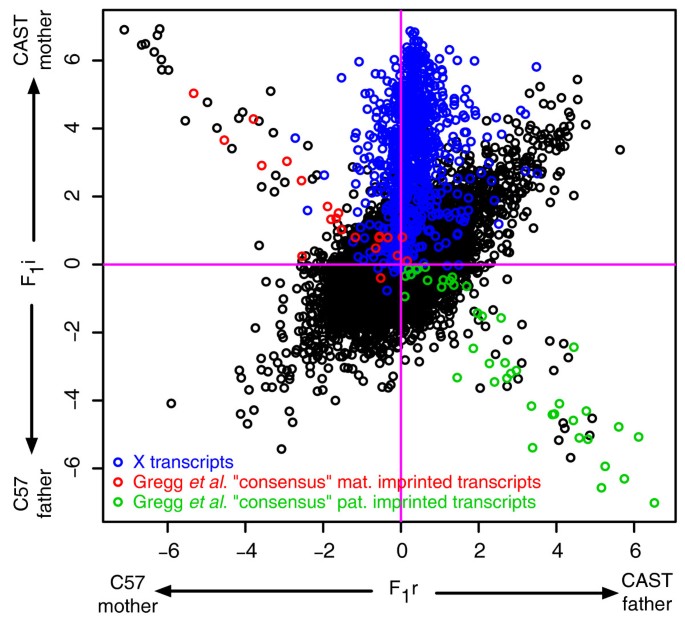
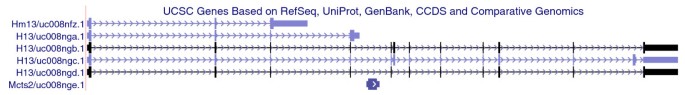
实验目的是验证MMSEQ在解析基因组印记、顺式调控等生物学问题中的实用性;方法细节:分析CAST/EiJ与C57BL/6J小鼠的F1杂交胚胎脑组织RNA测序数据,比较初始杂交与 reciprocal 杂交的单倍型-转录本异构体表达差异;结果解读:MMSEQ检测到三类转录本:一是基因组印记转录本,表达差异依赖于亲本来源(母源印记或父源印记);二是X染色体转录本,初始杂交中CAST单倍型表达更高,reciprocal杂交中表达平衡,提示初始杂交为雄性、reciprocal杂交为雌性;三是顺式调控转录本,表达差异不依赖于亲本来源;还发现同一基因不同转录本异构体的相反等位基因表达不平衡,如H13基因的短转录本异构体偏向父源表达,长转录本异构体偏向母源表达,说明转录本水平分析的必要性;产品关联:文献未提及具体实验产品,领域常规使用小鼠杂交模型的测序数据。
4. Biomarker研究及发现成果解析
本研究中涉及的生物标志物为单倍型特异性的转录本异构体表达不平衡,该标志物可用于解析基因组印记、顺式调控等生物学过程,其筛选与验证基于MMSEQ的表达估计结果与多组学数据的关联分析。
Biomarker定位:本研究定义的生物标志物为“单倍型-转录本异构体表达不平衡”,属于转录组水平的功能标志物,其筛选与验证逻辑为:首先通过MMSEQ估计单倍型与转录本异构体的表达水平,然后比较不同遗传背景样本(如小鼠初始杂交与reciprocal杂交样本)的表达差异,验证其与基因组印记、顺式调控的关联,最后通过模拟数据与真实数据验证标志物的可靠性。
研究过程详述:该生物标志物的来源为小鼠F1杂交胚胎脑组织的RNA测序样本(每个杂交组为4个个体的混合样本,n=4),验证方法为MMSEQ估计单倍型-转录本异构体的表达水平,计算表达 fold change,通过散点图聚类分析区分不同调控类型的转录本;特异性方面,该标志物能准确区分基因组印记转录本(亲本来源依赖)与顺式调控转录本(亲本来源无关),还能检测同一基因不同转录本异构体的相反调控模式;敏感性方面,即使在低表达转录本中,通过Gibbs抽样的正则化估计也能稳定检测到表达不平衡,无具体ROC曲线数据,但通过表达估计的Monte Carlo标准误(MCSE)验证了结果的稳定性。
核心成果提炼:该生物标志物的核心功能关联在于能直接反映转录本异构体水平的顺式调控与基因组印记效应,创新性在于首次实现了单倍型与转录本异构体水平的表达不平衡分析,解决了传统基于SNP分析的局限性,能揭示同一基因不同转录本异构体的相反调控模式;统计学结果显示,在小鼠数据中检测到27个转录本存在相反方向的SNP表达不平衡,说明仅基于SNP的分析会误导对转录本水平调控的判断,而本研究的标志物能提供更精准的调控信息;该成果为功能基因组学研究提供了新的分析工具,可应用于疾病相关的顺式调控变异、基因组印记异常等研究领域。