1. 领域背景与文献引入
文献英文标题:Predicting specificity in bZIP coiled-coil protein interactions;发表期刊:Genome Biology;影响因子:未公开;研究领域:计算生物学与蛋白质相互作用(bZIP转录因子卷曲螺旋相互作用特异性预测)
蛋白质相互作用是细胞信号传导、基因表达调控等生命活动的核心分子基础,卷曲螺旋(coiled-coil)是自然界中最常见的蛋白质相互作用基序之一,广泛存在于转录因子、细胞骨架蛋白、膜融合蛋白中,参与肿瘤发生、神经发育等关键生理病理过程。领域共识:1991年Lupas等人首次开发出基于序列的卷曲螺旋预测工具,开启了卷曲螺旋生物信息学研究的先河;后续陆续出现Multicoil、Learncoil-VMF等工具,可有效识别蛋白质序列中的卷曲螺旋区域,但针对天然卷曲螺旋蛋白的结合特异性预测,长期以来进展有限。早期研究中,部分方法仅能排除给定卷曲螺旋序列的非相互作用伙伴,无法精准定位实际结合伴侣;基于静电相互作用或耦合能的简单规则虽能做出部分预测,但存在假阳性率高、识别范围有限的问题,尤其在基因组尺度的大规模预测中,难以兼顾敏感性与特异性。
bZIP转录因子是一类包含亮氨酸拉链(即卷曲螺旋区域)的转录调控蛋白,其同源或异源二聚化的特异性直接决定了下游靶基因的选择与调控功能,是细胞命运决定、应激响应等过程的关键调控节点。针对bZIP蛋白相互作用特异性的精准预测,不仅能解析转录调控的分子机制,还能为疾病相关的功能突变研究提供线索。然而,现有方法在人类bZIP蛋白数据集上的表现差强人意:当设定阈值以识别至少1/3的已知强相互作用时,假阳性数量与真阳性数量相当,无法满足后续功能研究的需求。在此背景下,本研究旨在开发一种基于序列残基相互作用权重优化的计算方法,实现bZIP蛋白卷曲螺旋相互作用特异性的高置信度大规模预测,填补领域内针对特定蛋白相互作用基序的精准预测方法空白。
2. 文献综述解析
作者按方法技术路线的差异,将领域内现有研究分为三类:早期卷曲螺旋序列预测工具、基于残基相互作用规则的特异性预测方法、全基因组尺度的蛋白质相互作用预测方法,系统对比各类方法的技术优势、局限性及在bZIP蛋白数据集上的性能表现。
早期卷曲螺旋序列预测工具以Multicoil、Learncoil-VMF为代表,这类方法基于序列的七肽重复特征(abcdefg),能有效识别蛋白质序列中的卷曲螺旋区域,为后续相互作用研究提供候选对象,但核心局限性在于仅能判断序列是否具有形成卷曲螺旋的潜力,无法预测其与其他蛋白的结合特异性,无法满足功能调控机制研究的需求。基于残基相互作用规则的方法又可细分为两类:一类是基于ge"位置静电相互作用的简单规则,这类方法通过统计有利/不利的静电相互作用数量做出预测,但在人类bZIP蛋白数据集上,当识别1/3的已知强相互作用时,假阳性率高达50%;另一类是基于实验测定耦合能的方法,虽能部分反映残基相互作用的热力学特性,但同样存在假阳性率高的问题,且覆盖的残基相互作用类型有限。全基因组尺度的蛋白质相互作用预测方法,如基于基因共线性、基因融合事件的方法,能在基因组层面预测潜在的蛋白相互作用对,但这类方法针对特定相互作用基序的预测精度较低,无法实现对bZIP这类依赖二聚化特异性发挥功能的蛋白的精准预测。
通过对比现有研究的未解决问题,本研究的创新价值凸显:现有方法均无法实现对bZIP蛋白卷曲螺旋相互作用的大规模高置信度预测,而本研究首次将支持向量机(SVM)的权重优化框架引入卷曲螺旋相互作用预测,结合基础数据集与实验验证的bZIP相互作用数据,实现了70%强相互作用识别率与92%预测准确率的平衡,是领域内首个针对特定蛋白相互作用基序的大规模精准预测方法,为其他类型蛋白相互作用基序的预测提供了可借鉴的技术范式。
3. 研究思路总结与详细解析
本研究的核心目标是开发并验证一种基于序列残基相互作用权重优化的计算方法,实现bZIP蛋白卷曲螺旋相互作用特异性的高置信度预测;核心科学问题是如何通过量化卷曲螺旋核心位置的残基间相互作用,精准区分bZIP蛋白的相互作用对与非相互作用对;技术路线遵循“模型构建→数据集验证→交叉验证→机制解析”的闭环逻辑,从数学建模到实验数据验证,逐步验证方法的性能与泛化能力。
3.1 卷曲螺旋相互作用的数学建模与权重优化
实验目的:建立基于卷曲螺旋核心残基相互作用的量化评分框架,通过权重优化提升对相互作用与非相互作用对的区分能力。
方法细节:将二聚体卷曲螺旋的a、d、e、g核心位置的7种两两残基相互作用(包括a_i-d"i、d_i-a"{i+1}等)转化为2800维特征向量,向量的每个维度对应一种残基对在特定相互作用位置的出现次数;采用支持向量机(SVM)框架,结合包含已知卷曲螺旋序列、非相互作用序列(通过错位对齐已知相互作用链构建)、实验测定的卷曲螺旋相对稳定性数据的基础数据集,构建约束条件以优化权重向量;同时引入两种现有方法作为对照:基于简单静电规则的权重、基于实验测定耦合能的权重。
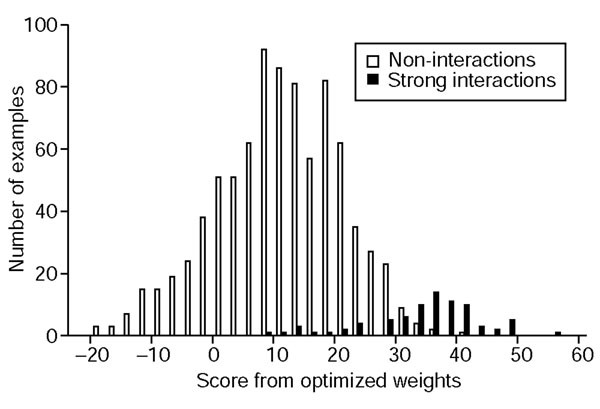
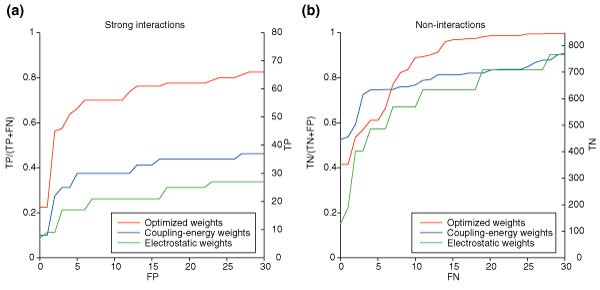
结果解读:基础优化权重的得分分布显示,强相互作用对的得分显著高于非相互作用对(图2),虽存在部分重叠,但整体区分度明显;ROC曲线分析表明,基础优化权重在识别强相互作用时的性能显著优于简单静电权重与耦合能权重(图3),当选取高置信度阈值时,可识别56个强相互作用(占实验测定强相互作用的70%),仅产生5个假阳性,预测准确率达92%;而简单静电权重在识别27个强相互作用时,会产生31个假阳性,预测精度仅46.6%。
产品关联:文献未提及具体实验产品,领域常规使用支持向量机工具包(如SVM-lite)、生物信息学分析软件进行序列建模与权重优化。
3.2 人类与酵母bZIP数据集的大规模性能验证
实验目的:在经过蛋白芯片实验验证的大规模bZIP相互作用数据集上,系统评估方法的预测精度与普适性。
方法细节:采用Newman等人2003年发表的蛋白芯片实验数据,筛选得到58个非冗余的人类与酵母bZIP蛋白;基于实验测定的Z-score,定义80个强相互作用(双向Z-score均>10)与849个非相互作用(双向Z-score均<1);分别用三种权重方法对所有蛋白对进行评分,计算真阳性(TP)、真阴性(TN)、假阳性(FP)、假阴性(FN)的数量,评估预测的敏感性、特异性与精度。
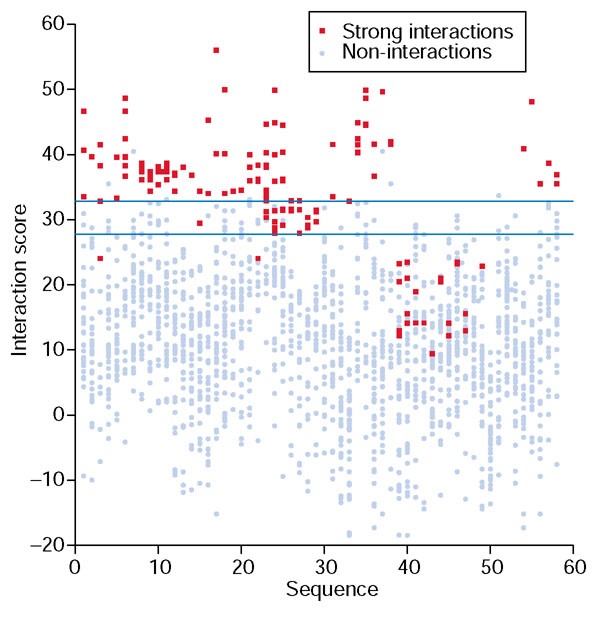
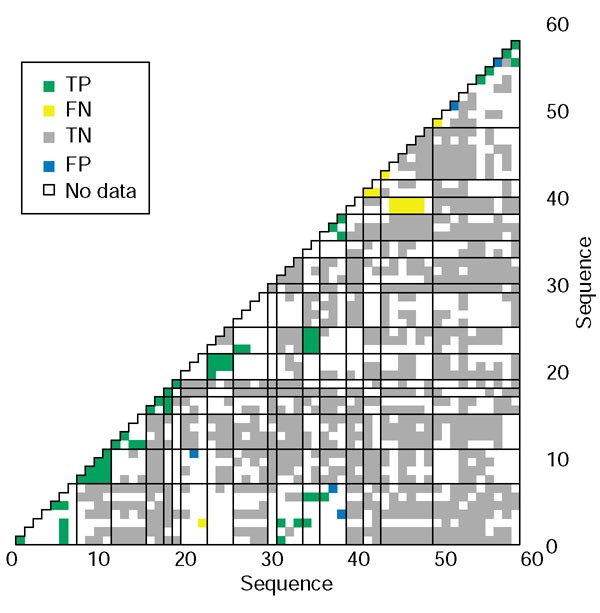
结果解读:基础优化权重在非相互作用预测中的表现同样优异,可排除89%的非相互作用对,同时保留83%的强相互作用,阴性预测值达98.4%(图3b);图4的蛋白水平得分分布显示,几乎所有蛋白的强相互作用得分(红色)均高于非相互作用得分(蓝色),说明方法对不同家族的bZIP蛋白均具有普适性;图5的网格图直观展示了所有蛋白对的预测结果,正确预测的强相互作用(绿色)与非相互作用(灰色)占比极高,不仅能准确识别家族内的同源二聚化,还能精准预测跨家族的异源二聚化相互作用。
产品关联:文献未提及具体实验产品,领域常规使用蛋白芯片平台、荧光定量检测系统获取bZIP相互作用实验数据。
3.3 交叉验证与方法泛化能力评估
实验目的:评估方法在新基因组bZIP蛋白中的预测能力,验证人类bZIP相互作用数据对模型优化的贡献,为跨物种预测提供依据。
方法细节:采用留一法交叉验证策略,每次排除一个bZIP家族,用剩余家族的bZIP数据优化权重向量,对排除家族的蛋白进行相互作用预测;计算每个序列的CV-similarity(即与其他家族bZIP蛋白在a、d、g、e位置的最大序列一致性),将序列分为CV-similarity≥50%与<50%两组,分别评估性能;同时对比基于序列相似性的简单预测方法(直接将同源蛋白的相互作用对作为预测结果)。
结果解读:交叉验证结果显示,加入人类bZIP数据优化后,模型的预测性能显著提升(图7),CV-similarity≥50%的序列性能提升尤为明显;基于序列相似性的方法仅能识别30.5%的可能相互作用(允许3.1个假阳性),而本研究的方法在无假阳性时能识别53.1%的相互作用,允许3个假阳性时能识别66.2%,性能远超简单同源预测方法,说明模型并非依赖同源序列的简单迁移,而是真正学习到了卷曲螺旋相互作用的特异性编码规则。
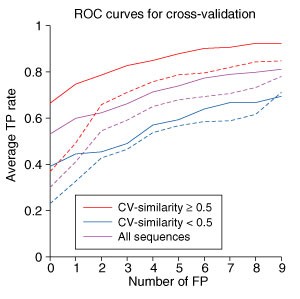
产品关联:文献未提及具体实验产品,领域常规使用序列比对工具(如BLAST)计算序列一致性,进行交叉验证分析。
4. Biomarker研究及发现成果解析
本研究中涉及的Biomarker为bZIP蛋白卷曲螺旋区域a、d、e、g核心位置的残基相互作用模式,作为预测bZIP蛋白二聚化特异性的分子标记,其筛选与验证遵循“序列特征提取→实验数据训练→交叉验证泛化”的完整逻辑链条。
该Biomarker属于功能型分子标记,定位为bZIP蛋白卷曲螺旋核心区域的残基组合与相互作用模式;筛选逻辑为:基于已知的bZIP相互作用实验数据,通过支持向量机优化权重,识别与相互作用特异性显著相关的残基间相互作用类型(包括dd"、da"等此前被忽略的相互作用);验证逻辑为:在人类与酵母bZIP数据集上验证其区分相互作用与非相互作用对的能力,通过交叉验证评估其在不同序列相似性群体中的泛化性能。
该Biomarker的来源为人类与酵母bZIP蛋白的卷曲螺旋核心区域序列;验证方法为计算每个蛋白对的相互作用得分,结合实验测定的Z-score评估标记的特异性与敏感性:在高置信度阈值下,标记的敏感性为70%(56/80,n=80),特异性为99.4%((849-5)/849,n=849);交叉验证中,CV-similarity≥50%的序列群体中,标记的预测敏感性更高,说明序列相似性可作为标记预测置信度的辅助指标;ROC曲线分析显示,基础优化权重的预测性能显著优于现有方法(文献未明确提供AUC值,基于图表趋势推测AUC>0.9)。
该残基相互作用模式作为bZIP蛋白二聚化特异性的功能标记,首次揭示了除ge"静电相互作用与aa"疏水相互作用外,dd"、da"等5种残基相互作用对bZIP蛋白结合特异性的关键调控作用;统计学结果显示,基于该标记的预测准确率达92%(56/61,n=61),交叉验证中无假阳性时的真阳性率为53.1%(文献未明确提供P值);创新性在于首次实现了bZIP蛋白相互作用的大规模高置信度预测,为其他物种bZIP蛋白的相互作用预测提供了可量化的标记(序列一致性与相互作用得分),同时为解析卷曲螺旋相互作用的特异性编码机制提供了新的视角。