1. 领域背景与文献引入
文献英文标题:Protein kinase C activation disrupts epithelial apical junctions via ROCK-II dependent stimulation of actomyosin contractility;发表期刊:BMC Cell Biology;影响因子:未公开;研究领域:肿瘤细胞生物学(上皮间质转化与细胞连接调控)
上皮间质转化(EMT)是肿瘤细胞获得侵袭和转移能力的核心生物学过程,其中上皮细胞间紧密连接(TJs)、黏着连接(AJs)的解离是EMT的标志性事件之一,直接驱动肿瘤细胞从原发灶脱离。领域发展关键节点包括:1980年代EMT概念正式提出,1990年代蛋白激酶C(PKC)被确认为肿瘤促进剂的核心靶点,2000年代非肌球蛋白II(NM II)在细胞连接调控中的作用逐步阐明。当前研究热点聚焦于EMT的分子调控网络,尤其是激酶信号通路对上皮细胞连接的精准调控机制,未解决的核心问题包括:PKC激活诱导上皮连接解离的具体下游信号通路、NM II的激活是否依赖经典RhoA信号、Rho相关激酶(ROCK)亚型在该过程中的特异性功能。结合领域现状,现有研究虽已证实PKC激活可诱导多种上皮细胞的连接解离,但针对人类肿瘤细胞系的机制研究仍存在空白,尤其是ROCK亚型的作用及RhoA的依赖性尚未明确。本研究以HPAF-II人胰腺腺癌细胞系为模型,解析PKC激活诱导上皮顶端连接解离的分子机制,为肿瘤转移的干预提供新的靶点依据。
2. 文献综述解析
作者从信号通路特异性、细胞模型差异、分子调控层级三个维度对现有研究进行分类评述,系统梳理了PKC激活诱导上皮连接解离的研究进展,明确了现有研究的核心结论、技术优势与局限性,并提出本研究的创新切入点。
现有研究的关键结论包括:PKC激活剂(如佛波酯TPA)可诱导多种上皮细胞系的TJs和AJs解离,NM II的收缩活性是调控上皮细胞连接稳定性的核心动力,ROCK和肌球蛋白轻链激酶(MLCK)是介导NM II激活的关键激酶。技术方法上,现有研究多采用药理学抑制剂结合免疫荧光、跨上皮电阻(TEER)检测等技术,能直观反映细胞连接的功能与结构变化,但也存在局限性:多数研究基于非人类或非肿瘤细胞系,结果的肿瘤相关性不足;对PKC下游调控NM II的具体激酶通路解析不充分,尤其是ROCK亚型的特异性作用未被阐明;且未明确该过程是否依赖经典RhoA信号。通过对比现有研究的空白,本研究的创新价值在于首次在人类胰腺肿瘤细胞系中证实经典PKC亚型介导上皮连接解离,首次明确ROCK-II而非ROCK-I在PKC激活诱导NM II激活中的特异性作用,且该过程不依赖RhoA信号,完善了PKC诱导EMT的分子调控网络,为肿瘤转移的机制研究提供了新的实验证据。
3. 研究思路总结与详细解析
本研究的整体框架以“PKC激活→信号通路筛选→NM II激活验证→ROCK亚型特异性分析→非依赖RhoA机制确认”为闭环,核心科学问题是PKC激活如何通过下游信号通路调控NM II的收缩活性,进而诱导上皮顶端连接解离,研究目标是明确该过程中的关键分子与信号节点。
3.1 上皮细胞单层模型构建与PKC激活剂功能验证
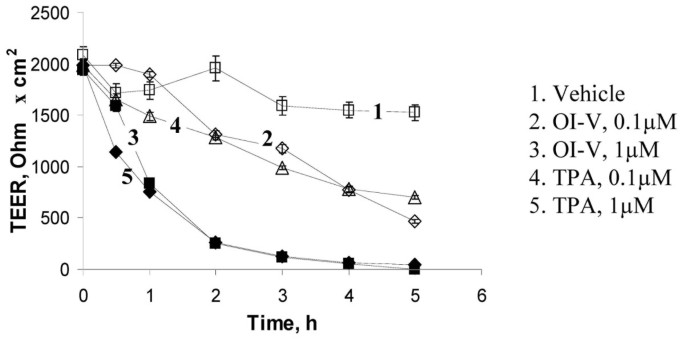
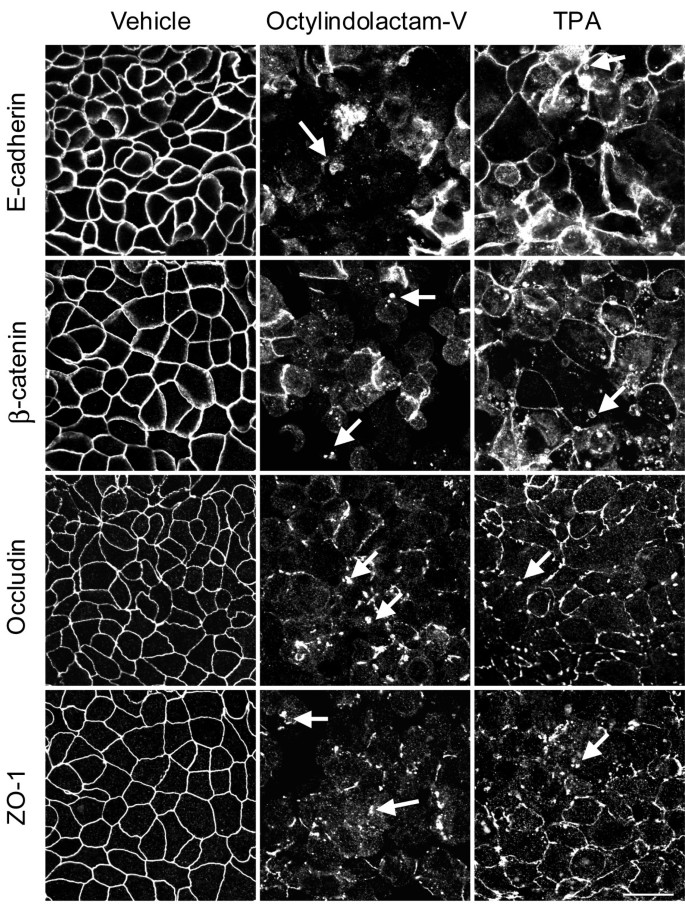
实验目的:验证PKC激活剂对HPAF-II胰腺腺癌细胞上皮连接功能与结构的影响,建立实验模型。方法细节:将HPAF-II细胞接种于胶原包被的Transwell滤膜上,培养6-10天形成高电阻上皮单层(跨上皮电阻(TEER)为1500-2100 Ohm×cm²);分别用0.1μM、1μM的octylindolactam-V(OI-V)或TPA处理细胞5小时,同时设置溶剂对照组,通过TEER检测评估上皮屏障功能,采用免疫荧光染色结合激光共聚焦显微镜观察E-钙黏蛋白、β-连环蛋白、闭合蛋白、ZO-1的定位变化。结果解读:TEER检测结果显示,1μM OI-V或TPA处理后,细胞单层的TEER从初始约2000 Ohm×cm²降至约5 Ohm×cm²(n=3,P<0.01),而溶剂对照组TEER维持在1500 Ohm×cm²以上;免疫荧光结果显示,OI-V和TPA处理后,原本定位于细胞间的连接蛋白转移至胞质形成点状结构,其中OI-V的作用效果强于TPA,表明PKC激活剂可显著破坏上皮连接的功能与结构。产品关联:实验所用关键产品:Invitrogen的E-钙黏蛋白、闭合蛋白抗体,Sigma的OI-V、TPA试剂,Zeiss LSM510激光共聚焦显微镜。
3.2 经典PKC亚型介导连接解离的特异性验证
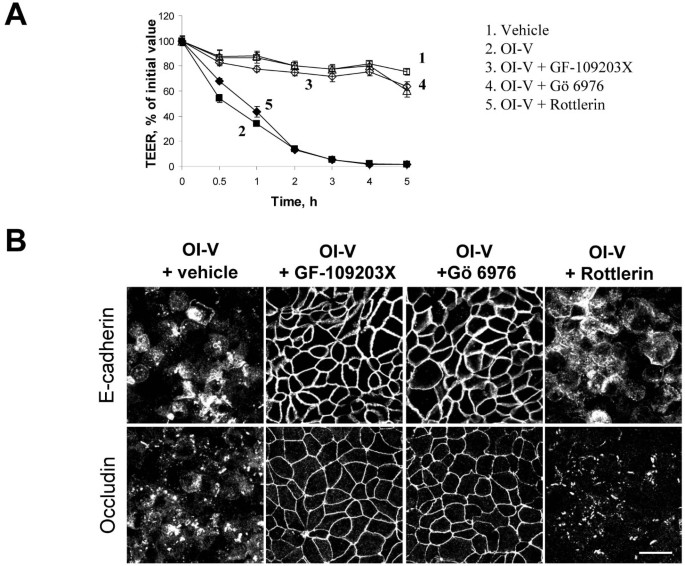
实验目的:确定参与PKC激活诱导上皮连接解离的PKC亚型类别。方法细节:采用RT-PCR和免疫印迹技术检测HPAF-II细胞中PKC亚型的表达谱;分别用广谱PKC抑制剂GF-109203X(10μM)、经典PKC抑制剂Gö 6976(5μM)、新型PKC抑制剂rottlerin(10μM)预处理细胞30分钟,再用1μM OI-V处理5小时,通过TEER检测和免疫荧光染色评估上皮连接的变化。结果解读:RT-PCR结果显示HPAF-II细胞表达经典PKCα、βI、γ和新型PKCδ、θ、ε、η;TEER检测和免疫荧光结果显示,GF-109203X和Gö 6976可显著抑制OI-V诱导的TEER下降和连接蛋白解离,而rottlerin无明显作用,表明经典PKC亚型是介导该过程的关键分子。产品关联:实验所用关键产品:Axxora LLC的GF-109203X,EMD Biosciences的Gö 6976,Sigma的rottlerin。
3.3 NM II在PKC诱导连接解离中的核心作用验证
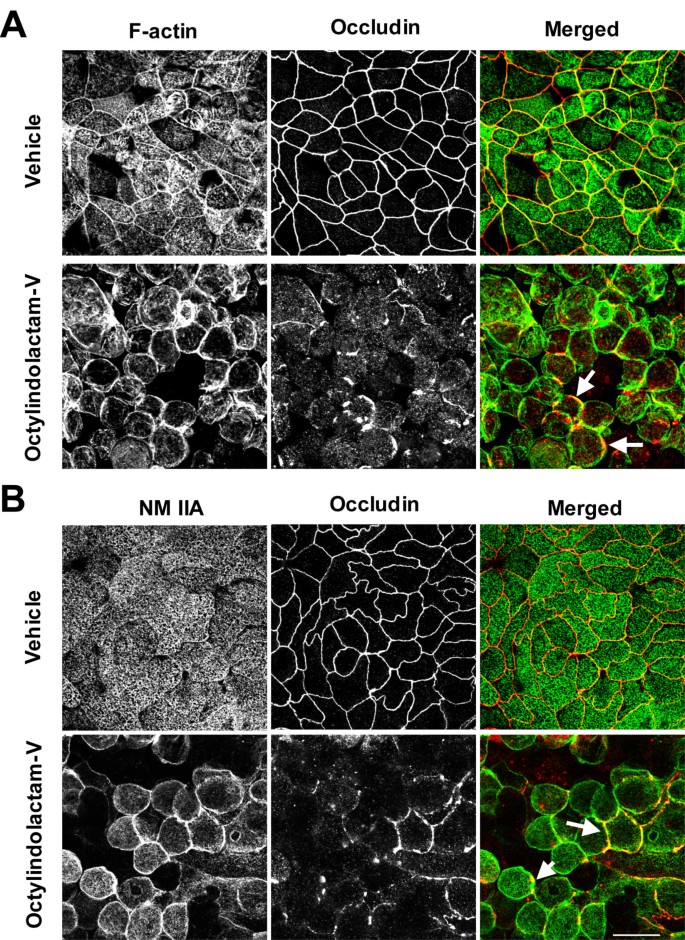
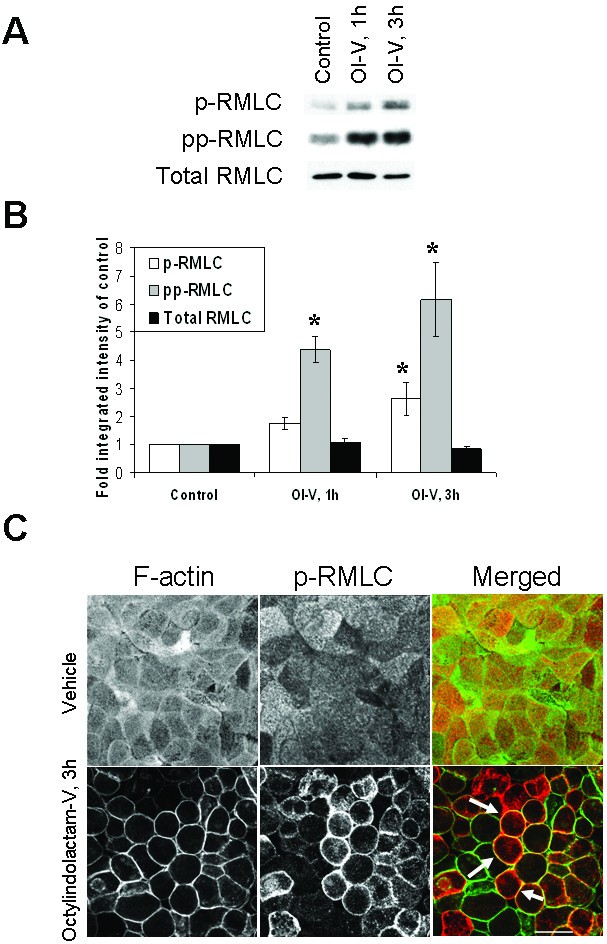
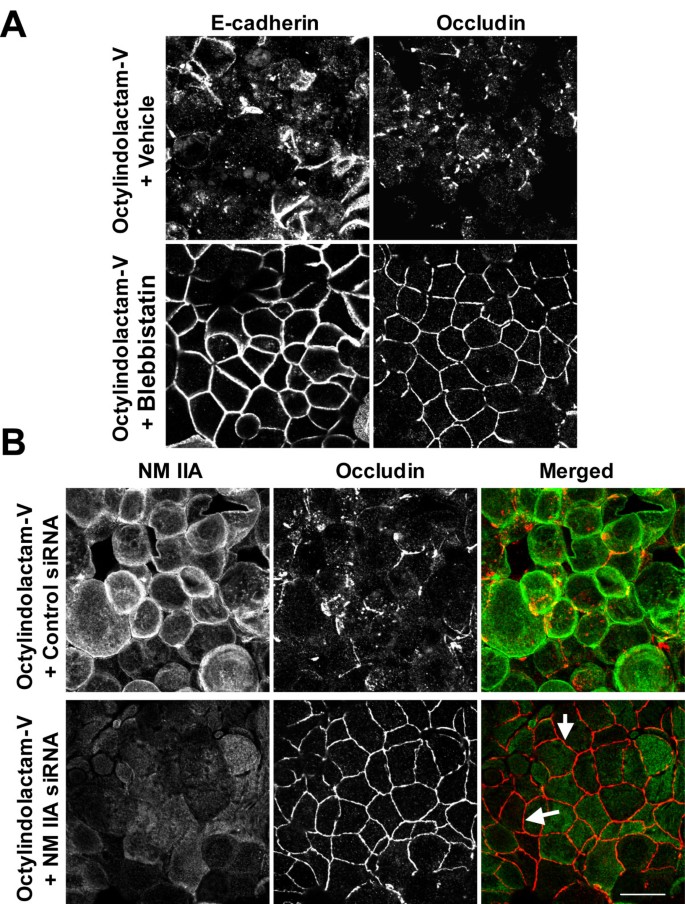
实验目的:探究NM II的激活与定位变化是否参与PKC诱导的上皮连接解离。方法细节:采用荧光标记的鬼笔环肽染色观察F-actin的定位变化,免疫荧光染色检测NM IIA的亚细胞定位;通过免疫印迹检测肌球蛋白轻链(RMLC)的单磷酸化和双磷酸化水平;分别用NM II抑制剂blebbistatin(100μM)预处理细胞,或用siRNA敲低NM IIA表达,再用OI-V处理,评估上皮连接结构的变化。结果解读:OI-V处理后,F-actin在细胞侧面聚集,NM IIA从顶端转移至细胞侧面的连接解离区域;免疫印迹结果显示,RMLC的单磷酸化和双磷酸化水平在处理后1-3小时显著升高(n=4,P<0.05),而总RMLC水平无变化;blebbistatin预处理和NM IIA siRNA敲低均可显著抑制OI-V诱导的连接蛋白解离,表明NM II的激活是PKC诱导连接解离的必要条件。产品关联:实验所用关键产品:Sigma的blebbistatin,Dharmacon的NM IIA siRNA,Cell Signaling Technology的磷酸化RMLC抗体。
3.4 ROCK-II介导PKC诱导的NM II激活验证
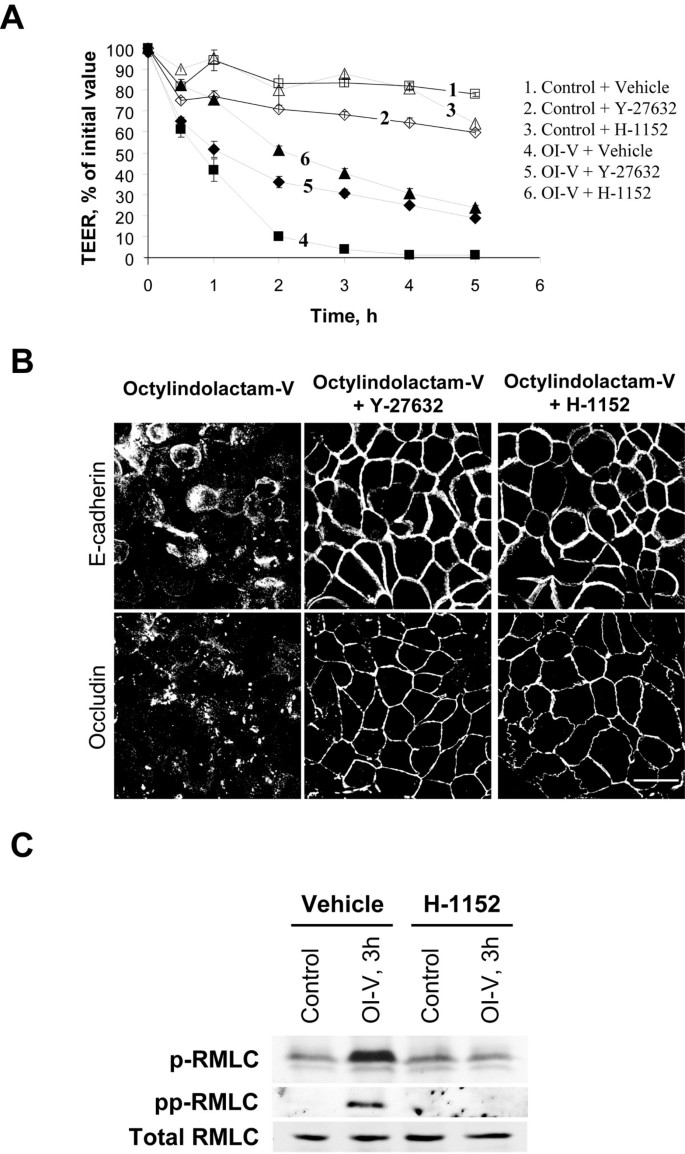
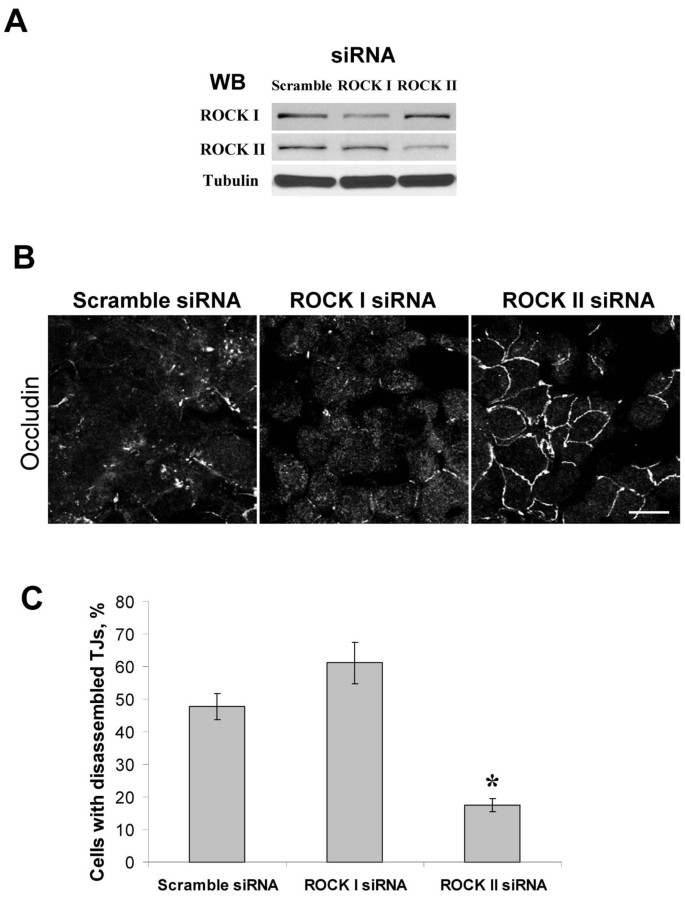
实验目的:确定调控NM II激活的下游激酶,并明确ROCK亚型的特异性作用。方法细节:采用ROCK抑制剂Y-27632(20μM)和H-1152(10μM)预处理细胞,检测OI-V处理后的TEER变化和连接结构,同时通过免疫印迹检测RMLC的磷酸化水平;用siRNA分别敲低ROCK-I和ROCK-II的表达,再用OI-V处理,评估上皮连接结构的变化。结果解读:Y-27632和H-1152均可显著抑制OI-V诱导的TEER下降和连接蛋白解离,并完全阻断RMLC的磷酸化;siRNA敲低结果显示,ROCK-II表达下调约70%(n=3,P<0.05)可显著抑制OI-V诱导的连接解离,而ROCK-I敲低无明显作用,表明ROCK-II是PKC下游激活NM II的关键激酶。产品关联:实验所用关键产品:EMD Biosciences的Y-27632、H-1152,QIAGEN的ROCK-I/II siRNA。
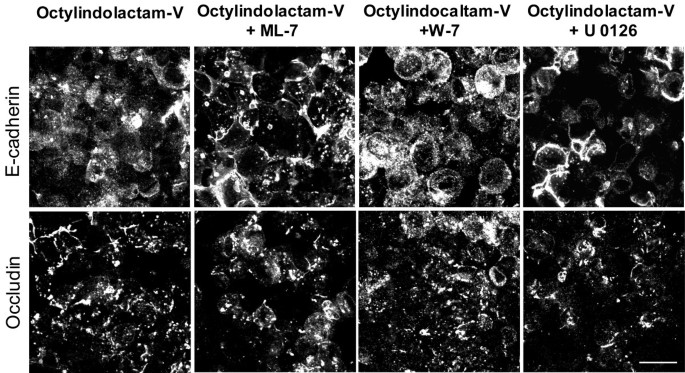
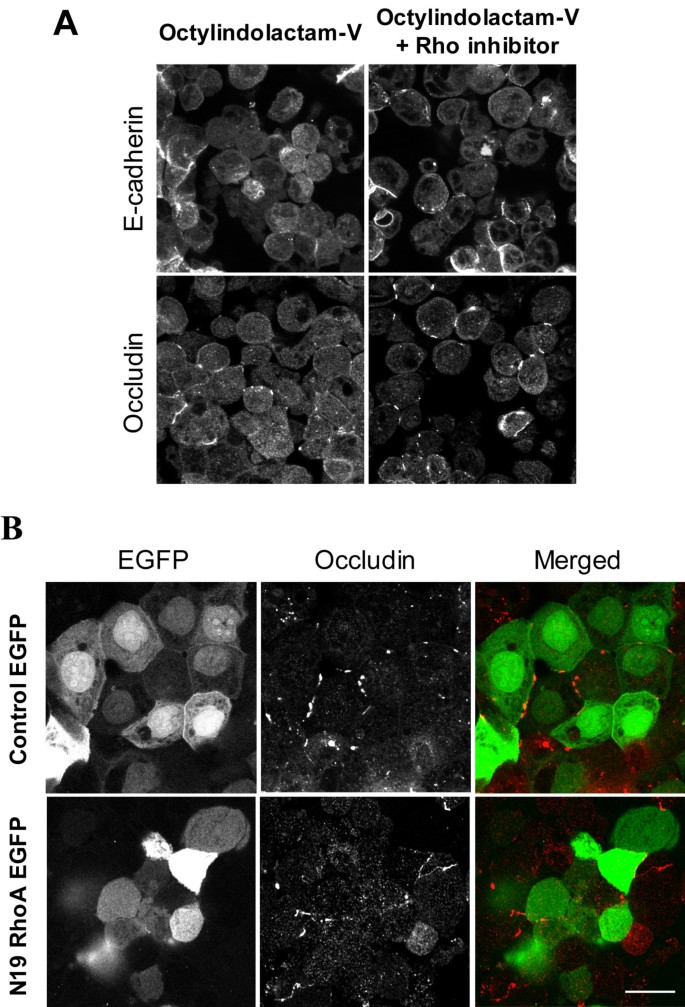
3.5 RhoA、MLCK、ERK1/2及凋亡的排除性验证
实验目的:验证PKC-ROCK-II通路是否依赖RhoA信号,以及其他激酶或凋亡是否参与该过程。方法细节:采用细胞渗透型Rho抑制剂C3毒素(2μg/ml)预处理细胞,或过表达显性负性RhoA(N19)突变体,检测OI-V处理后的连接结构变化;分别用MLCK抑制剂ML-7(20μM)、钙调蛋白抑制剂W-7(100μM)、ERK1/2抑制剂U0126(10μM)预处理细胞,评估OI-V诱导的连接解离变化;采用泛caspase抑制剂z-VAD-fmk(50μM)预处理细胞,同时检测caspase激活情况,验证凋亡是否参与。结果解读:Rho抑制剂和N19 RhoA过表达均无法抑制OI-V诱导的连接解离,ML-7、W-7、U0126也无明显抑制作用;z-VAD-fmk预处理对连接解离无影响,且OI-V处理未诱导caspase激活,表明PKC诱导的连接解离不依赖RhoA、MLCK、ERK1/2信号及凋亡途径。产品关联:实验所用关键产品:Cytoskeleton Inc.的C3毒素,Sigma的ML-7、W-7、U0126,Axxora LLC的z-VAD-fmk。
4. Biomarker研究及发现成果解析
本研究聚焦于功能型Biomarker的解析,明确ROCK-II是PKC激活诱导上皮连接解离的关键功能Biomarker,其筛选与验证遵循“药理学筛选→亚型特异性验证→非依赖机制确认”的完整逻辑链条。
Biomarker定位:ROCK-II属于丝氨酸/苏氨酸激酶,是PKC诱导肿瘤细胞EMT过程中的关键调控分子,筛选与验证逻辑为:首先通过ROCK抑制剂证明ROCK家族参与PKC诱导的连接解离,再通过siRNA敲低验证ROCK-II的特异性作用,最后排除RhoA依赖机制,明确其独立调控功能。研究过程详述:ROCK-II来源于HPAF-II细胞的内源性表达,验证方法包括药理学抑制剂处理和siRNA敲低,特异性数据显示,ROCK-II siRNA可使ROCK-II表达水平下调约70%(n=3,P<0.05),并将OI-V诱导的紧密连接解离细胞比例从约85%降至约20%(n=3,P<0.01),而ROCK-I siRNA无此作用;敏感性数据显示,H-1152(10μM)可完全抑制OI-V诱导的RMLC磷酸化(n=3,P<0.01)。核心成果提炼:ROCK-II作为PKC下游的关键激酶,通过激活NM II的收缩活性诱导上皮顶端连接解离,其功能关联是肿瘤细胞转移的潜在调控靶点,创新性在于首次证明ROCK-II在PKC诱导的EMT过程中的特异性作用,且该过程不依赖经典RhoA信号,为肿瘤转移的机制研究提供了新的分子靶点,也为抗肿瘤药物的研发提供了新的方向。