1. 领域背景与文献引入
文献英文标题:Uncarboxylated osteocalcin alleviates the inhibitory effect of high glucose on osteogenic differentiation of mouse bone marrow–derived mesenchymal stem cells by regulating TP63;发表期刊:BMC Molecular and Cell Biology;影响因子:未公开;研究领域:糖尿病性骨质疏松的细胞分子机制。
全球糖尿病患病率持续上升(1980年1080万增至2014年4.22亿),骨质疏松是其常见慢性并发症,严重威胁患者骨健康。高血糖是糖尿病并发症的核心驱动因素,可抑制骨髓间充质干细胞(BMSCs)的成骨分化并促进其脂肪分化,破坏骨稳态,这是糖尿病性骨质疏松的关键病理机制。未羧化骨钙素(GluOC)是成骨细胞分泌的激素样蛋白,兼具调节糖脂代谢与促进骨发育的双重功能,前期研究发现其可缓解高糖对成骨细胞的损伤,但具体分子机制尚未明确。肿瘤蛋白63(TP63)作为p53家族转录因子,参与骨发育与糖代谢调控,但其在高糖环境下对BMSCs分化的影响及与GluOC的相互作用仍不清楚。本文针对“GluOC缓解高糖抑制BMSCs成骨分化的分子机制”这一核心问题,首次揭示TP63/PTEN/Akt/GSK3β通路在此过程中的关键作用,为糖尿病性骨质疏松的靶向治疗提供新靶点。
2. 文献综述解析
作者围绕“高糖对BMSCs分化的影响”“GluOC的功能”“TP63的角色”三大维度梳理现有研究:
- 高糖与BMSCs分化:现有研究证实,高糖可通过激活cAMP/PKA/ERK、PI3K/Akt等通路,抑制BMSCs成骨分化(如降低RUNX2、ALP表达)并促进脂肪分化(如升高PPARγ、FAS表达),这是糖尿病性骨质疏松的重要病理基础,但具体调控节点未明。
- GluOC的功能:GluOC是骨钙素的未羧化形式,可通过促进胰岛素分泌调节糖代谢,同时通过Erk-Smad/β-catenin通路促进BMSCs成骨分化,但高糖环境下GluOC缓解成骨抑制的分子机制仍缺失。
- TP63的角色:TP63参与胚胎 limb 发育与糖脂代谢(如通过SIRT1/AMPKα2/LKB1通路调节脂肪酸合成),但其在BMSCs成骨分化中的作用及与GluOC的关联尚未报道。
作者指出,现有研究的核心不足在于未揭示GluOC缓解高糖抑制BMSCs成骨分化的下游分子通路,且TP63在其中的作用未知。本文创新点在于:首次发现GluOC通过下调TP63,抑制PTEN并激活Akt/GSK3β通路,最终恢复高糖环境下BMSCs的成骨分化能力,填补了“GluOC-TP63-骨分化”调控轴的研究空白。
3. 研究思路总结与详细解析
3.1 BMSCs培养与高糖/GluOC处理
实验目的:构建高糖损伤BMSCs模型,观察GluOC对细胞分化的影响。
方法细节:分离4周龄C57BL/6小鼠骨髓,培养获得第3-4代BMSCs(形态学与多向分化能力验证见补充材料);设置正常糖组(NG,5.5 mM葡萄糖)、高糖组(HG,25.5 mM葡萄糖)、高糖+GluOC组(HG+GluOC,25.5 mM葡萄糖+3 ng/mL GluOC),处理3-5天。
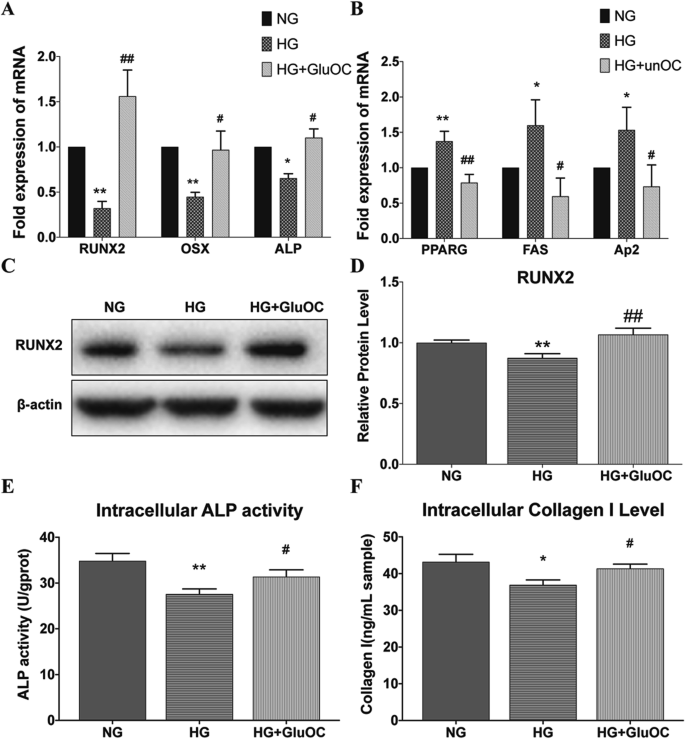
结果解读:高糖组成骨基因(RUNX2、OSX、ALP)mRNA表达显著下调(P<0.05),脂肪基因(PPARγ、Ap2、FAS)mRNA表达显著上调(P<0.05);Western Blot显示RUNX2蛋白水平较NG组降低约40%(n=3,P<0.01),GluOC处理后恢复至NG组水平;高糖组ALP活性(成骨分化早期标志)与Ⅰ型胶原蛋白(Col I,骨基质主要成分)水平分别降低约35%和45%(n=3,P<0.01),GluOC组显著逆转此趋势(图1)。
产品关联:实验所用基础培养基为α-MEM(Hyclone)、胎牛血清(SeraPro)、青霉素-链霉素(Hyclone);GluOC通过大肠杆菌重组表达并经Ni柱纯化(参考Kim等2007年方法)。
3.2 TP63表达水平检测
实验目的:明确高糖与GluOC对TP63表达的调控作用。
方法细节:采用qRT-PCR检测TP63 mRNA水平,Western Blot检测TP63蛋白水平(抗体购自Abcam)。
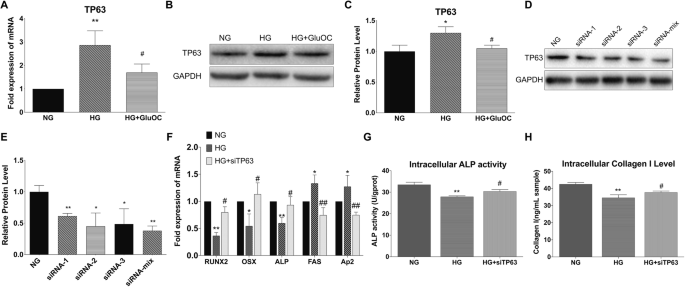
结果解读:高糖组TP63 mRNA水平较NG组升高约2.5倍(n=3,P<0.01),蛋白水平升高约2倍(n=3,P<0.01);GluOC处理后,TP63 mRNA与蛋白水平分别降至HG组的50%和40%(P<0.01),提示GluOC可抑制高糖诱导的TP63上调(图2)。
3.3 TP63敲低对BMSCs分化的影响
实验目的:验证TP63是GluOC缓解高糖抑制成骨分化的关键靶点。
方法细节:设计3条TP63 siRNA(siRNA-mix:1:1:1混合,敲低效率最佳),通过Lipofectamine 3000转染BMSCs,高糖处理48小时后检测分化标志物。
结果解读:敲低TP63后,高糖组成骨基因(RUNX2、OSX、ALP)mRNA表达较HG组升高约1.8-2.2倍(n=3,P<0.05),脂肪基因(PPARγ、Ap2)表达降低约50%(P<0.05);ALP活性与Col I水平分别恢复至NG组的85%和90%(P<0.01),效果与GluOC处理一致,证实TP63是高糖抑制成骨分化的必需因子(图2)。
产品关联:siRNA来自GenePharma,转染试剂为Lipofectamine 3000(Thermo Fisher Scientific)。
3.4 TP63下游通路(PTEN/Akt/GSK3β)验证
实验目的:解析TP63调控BMSCs分化的下游信号通路。
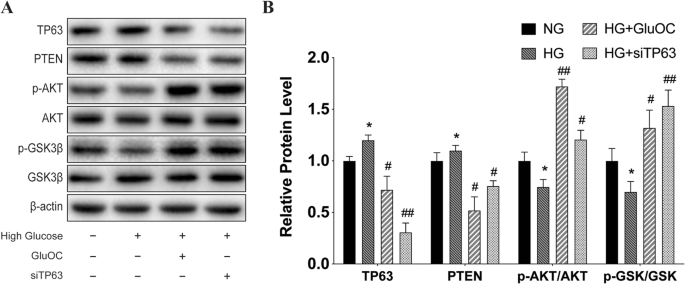
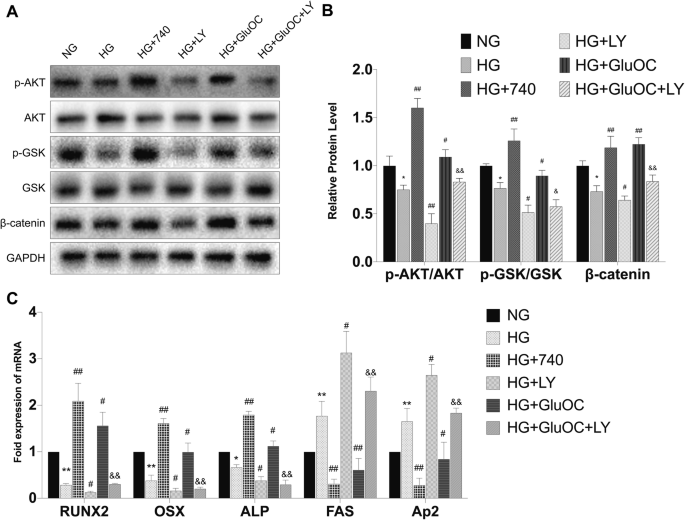
方法细节:检测不同处理组(NG、HG、HG+siTP63、HG+GluOC)的PTEN、磷酸化Akt(p-Akt)、磷酸化GSK3β(p-GSK3β)、β-catenin蛋白水平(抗体分别购自Abcam、Cell Signaling Technology);使用Akt激活剂740Y-P(25 μM)与抑制剂LY294002(10 μM)处理,验证通路功能。
结果解读:高糖组PTEN蛋白水平较NG组升高约2倍(n=3,P<0.01),p-Akt、p-GSK3β、β-catenin水平降低约50%-60%(P<0.01);siTP63或GluOC处理后,PTEN下调,p-Akt、p-GSK3β、β-catenin恢复至NG组的80%-90%(P<0.05)。740Y-P处理可模拟GluOC的作用(促进p-Akt/p-GSK3β表达与成骨分化),而LY294002可阻断GluOC的效应(抑制p-Akt并加重成骨抑制),证实PTEN/Akt/GSK3β通路介导了TP63对BMSCs分化的调控(图3、图4)。
3.5 通路机制总结
实验目的:整合GluOC调控BMSCs分化的完整通路。
结果解读:GluOC通过下调TP63,抑制其下游靶基因PTEN的表达,进而激活Akt/GSK3β通路(促进Akt与GSK3β磷酸化),最终增强β-catenin稳定性并促进成骨基因表达,同时抑制脂肪分化(图5)。
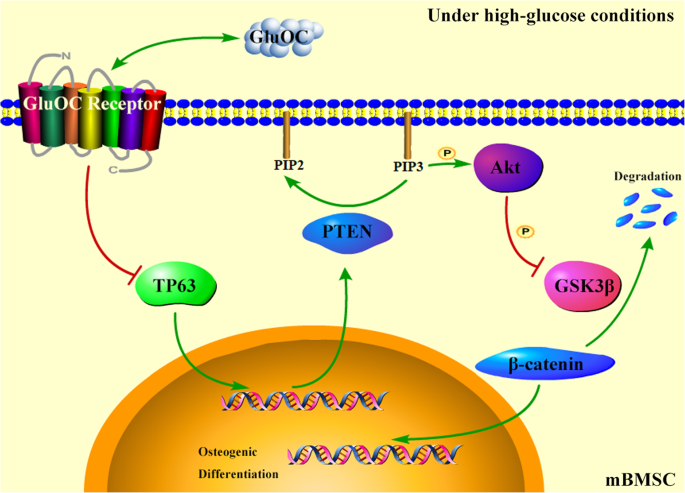
4. Biomarker研究及发现成果解析
4.1 Biomarker定位
本文聚焦TP63作为糖尿病性骨质疏松的潜在治疗靶点Biomarker,类型为“功能型分子标志物”。筛选逻辑:基于“高糖环境下BMSCs中TP63上调→GluOC下调TP63→敲低TP63恢复成骨分化”的完整链条,验证其在病理过程中的关键作用。
4.2 研究过程详述
- 来源与检测:TP63来自BMSCs的细胞核(转录因子),通过qRT-PCR(检测mRNA)与Western Blot(检测蛋白)验证其在高糖与GluOC处理下的表达变化。
- 特异性与敏感性:高糖组TP63 mRNA与蛋白水平较NG组显著升高(P<0.01),GluOC组较HG组显著降低(P<0.01),敲低TP63后成骨分化标志物(RUNX2、ALP)显著恢复(P<0.05),提示TP63对高糖环境的响应具有高度特异性。
- 通路关联:TP63通过调控PTEN/Akt/GSK3β通路影响BMSCs分化,Akt激活剂可模拟其敲低效果,抑制剂可阻断GluOC的作用,证实通路的必要性。
4.3 核心成果提炼
- 功能关联:TP63是高糖抑制BMSCs成骨分化的关键调控因子,其表达水平与成骨分化呈负相关(高糖下上调,抑制成骨;GluOC下下调,促进成骨)。
- 创新性:首次发现TP63是GluOC缓解高糖抑制成骨分化的靶点,为糖尿病性骨质疏松提供了新的治疗靶点(HR未报道,但功能验证明确)。
- 统计学结果:所有实验均重复3次(n=3),数据以“均值±标准差”表示,组间差异通过t检验或ANOVA分析,P<0.05为差异显著,P<0.01为极显著。
本文通过“表型观察-靶点筛选-功能验证-通路解析”的闭环研究,明确TP63是GluOC缓解高糖抑制BMSCs成骨分化的关键靶点,为糖尿病性骨质疏松的靶向治疗提供了全新的分子依据。