1. 领域背景与文献引入
文献英文标题:pan-Draft: automated reconstruction of species-representative metabolic models from multiple genomes;发表期刊:Genome Biology;影响因子:未明确提供;研究领域:微生物代谢组学、宏基因组学,聚焦宏基因组组装基因组(MAGs)的物种水平基因组尺度代谢模型(GEMs)构建。
基因组尺度代谢模型(GEMs)是解析微生物代谢功能、预测生态角色与工业应用潜力的核心工具,自2000年代初首个原核生物GEM构建以来,先后出现ModelSEED、CarveMe、gapseq等自动化工具,推动了GEMs在微生物组研究中的规模化应用。当前研究热点集中于利用宏基因组组装基因组(MAGs)构建未培养微生物的GEMs,以突破可培养微生物仅占全球微生物多样性极小部分的限制。但领域内未解决的核心问题是,MAGs普遍存在不完整性和污染,导致单MAG构建的GEMs存在大量代谢缺口,无法准确反映物种的真实代谢能力;现有GEM构建工具多依赖单基因组的同源性搜索,或需要完整参考基因组,无法有效利用MAGs的冗余信息弥补其缺陷。
针对上述问题,本研究开发了pan-Draft工具并整合到gapseq流程中,通过泛反应组分析利用同一物种多个MAGs的冗余遗传信息,构建高质量的物种水平GEMs(pan-GEMs),为未培养微生物的代谢功能解析提供可靠方法,具有重要的生态和工业应用价值。
2. 文献综述解析
作者对领域内现有研究的分类维度为GEM构建工具的技术策略,分为基于单基因组同源性的自动化工具、基于泛基因组的模型构建工具两类。
基于单基因组同源性的工具(如CarveMe、gapseq、RAVEN等)通过基因-蛋白-反应规则预测代谢网络,能快速构建GEMs,但受限于MAGs的不完整性,构建的模型存在大量缺口,无法准确反映物种代谢能力;部分工具虽引入缺口填充策略,但仍依赖单基因组信息,难以解决MAGs的污染问题。基于泛基因组的工具(如createPanModels、MIGRENE Toolbox)通过整合同一物种多个基因组构建泛模型,能提高模型的完整性,但createPanModels仅适用于AGORA数据库中的完整基因组,MIGRENE Toolbox依赖肠道微生物参考模型,应用场景受限,且无法有效处理无参考基因组的未培养物种。现有研究的局限性在于,缺乏不依赖参考基因组、能有效利用MAGs冗余信息解决其不完整性和污染问题的GEM构建方法。
本研究的创新点在于提出泛反应组分析策略,不依赖参考基因组,通过统计同一物种多个MAGs中代谢反应的出现频率,筛选出核心代谢反应构建pan-GEMs,同时利用反应频率优化缺口填充步骤,有效解决了MAGs不完整性和污染导致的模型质量问题;与现有泛基因组工具相比,pan-Draft具有更强的通用性,可应用于任何环境的原核生物MAGs,无需参考基因组,填补了无参考基因组未培养物种高质量GEM构建的空白。
3. 研究思路总结与详细解析
本研究的整体框架为:以解决MAGs构建GEMs的质量缺陷为核心科学问题,开发pan-Draft工具,通过泛反应组分析利用多MAGs冗余信息构建物种水平GEMs;技术路线为:筛选不同环境的MAGs数据集→分析MAG完整性与GEM质量的相关性→确定最优最小反应频率(MRF)阈值→构建pan-GEMs并评估其结构与功能质量→与现有工具进行性能比较,形成“问题提出→方法开发→验证优化→性能评估”的闭环逻辑。
3.1 数据集筛选与实验设计
实验目的:获取具有代表性的不同环境MAGs数据集,验证pan-Draft工具的通用性,同时明确未培养物种MAGs的质量现状。
方法细节:选取两个大型公共数据库——人类胃肠道基因组目录(UHGG v2.0.1)和海洋宏基因组数据库(OMD v1.1),筛选出包含至少30个MAGs的物种水平基因组箱(SGBs);采用dRep工具以99.9%平均核苷酸一致性(ANI)去除冗余基因组,保留完整性>50%、污染<5%的MAGs;同时筛选具有参考基因组的SGBs用于模型质量验证。
结果解读:最终得到75个UHGG的SGBs(包含62034个MAGs和4311个参考基因组)和9个OMD的SGBs(包含472个MAGs和16个参考基因组);统计显示,大部分SGBs无分离株参考基因组(UHGG中375个、OMD中126个),且部分SGBs的MAGs最高完整性不足90%,说明未培养物种MAGs的质量缺陷普遍存在,凸显了泛反应组策略的必要性。
产品关联:文献未提及具体实验产品,领域常规使用dRep进行基因组去冗余、GTDB-Tk进行物种分类、CheckM进行MAG质量评估。
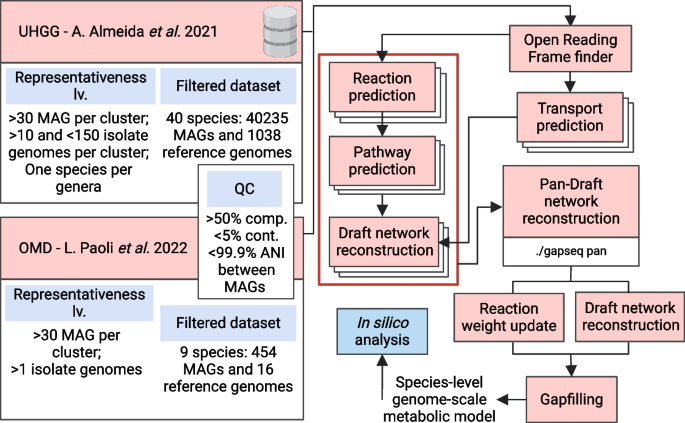
3.2 MAG完整性与GEM质量相关性分析
实验目的:明确单MAG构建GEMs的质量缺陷,为泛反应组策略的必要性提供依据。
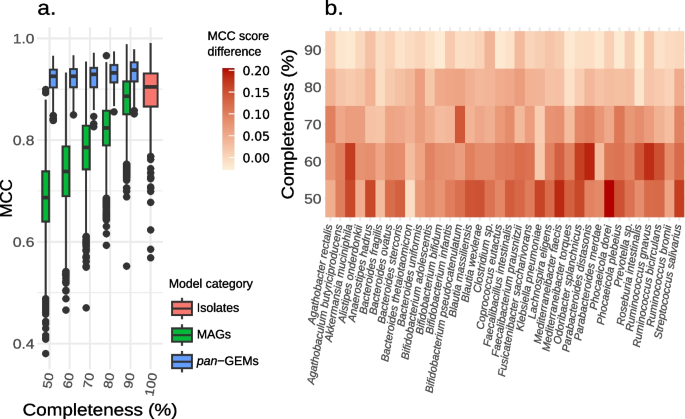
方法细节:利用gapseq工具构建单MAG的GEMs(MAG-GEMs)和参考基因组的GEMs(iso-GEMs);通过计算MAG-GEMs与iso-GEMs反应组的Matthews相关系数(MCC)评估模型质量;拟合广义加性模型分析MAG完整性与MCC的相关性。
结果解读:MAG完整性与MCC呈显著正相关(Spearman相关系数rho=0.78,P<2.2e-16),说明MAG完整性越低,构建的GEMs与参考模型的差异越大;部分未培养物种的MAGs最高完整性不足90%,其构建的GEMs存在大量代谢缺口,进一步验证了单MAG策略的局限性。
产品关联:文献未提及具体实验产品,领域常规使用gapseq构建GEMs、ggplot2进行统计可视化分析。
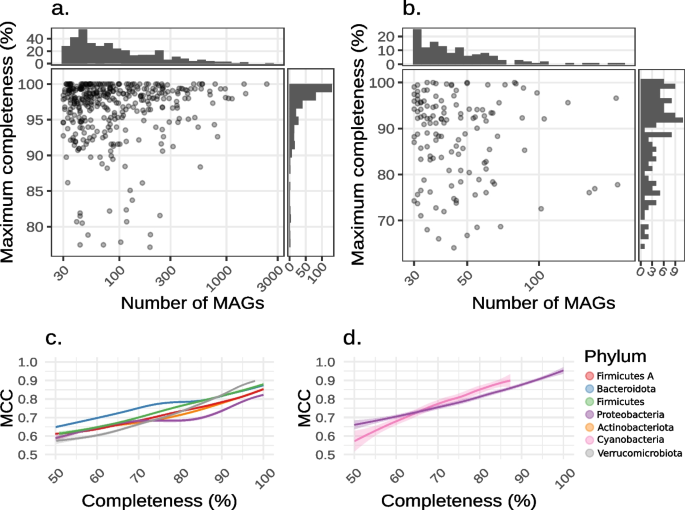
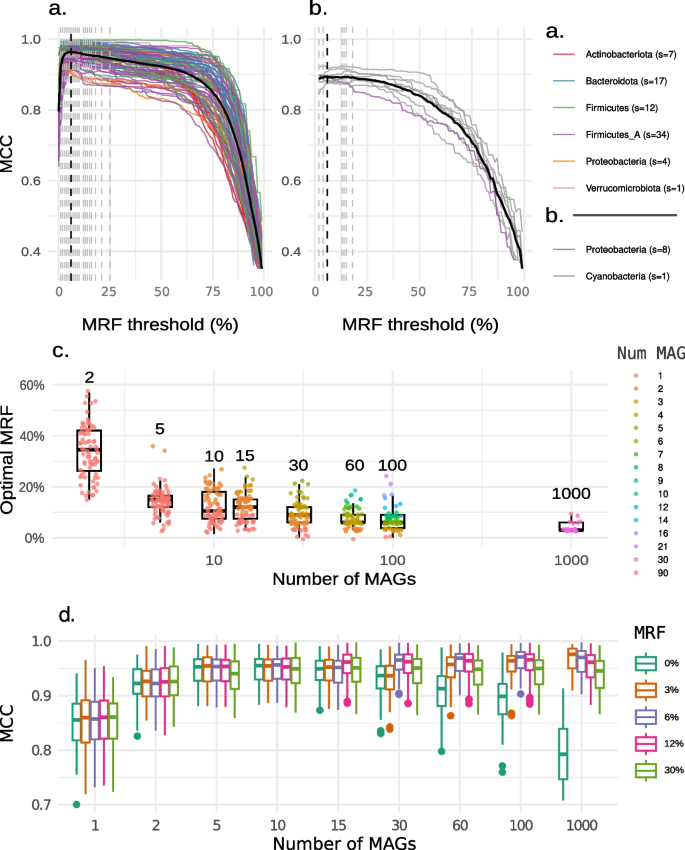
3.3 最小反应频率(MRF)阈值确定
实验目的:筛选最优的MRF阈值,平衡代谢反应的真实性(排除污染反应)和完整性(保留核心与附属代谢反应)。
方法细节:测试0-100%的MRF阈值,构建不同阈值下的pan-GEMs,计算其与iso-GEMs反应组的MCC;通过平均所有物种的MCC确定最优阈值;同时验证不同MAG数量下阈值的稳定性。
结果解读:最优MRF阈值为6%,该阈值下pan-GEMs与参考模型的MCC最高;当MAG数量≥15时,6%的阈值能有效排除污染反应,同时保留大部分核心和附属代谢反应;当MAG数量<15时,阈值可适当降低至接近1/n(n为MAG数量),以保留更多反应。
产品关联:文献未提及具体实验产品,领域常规使用R语言进行统计分析与阈值优化。
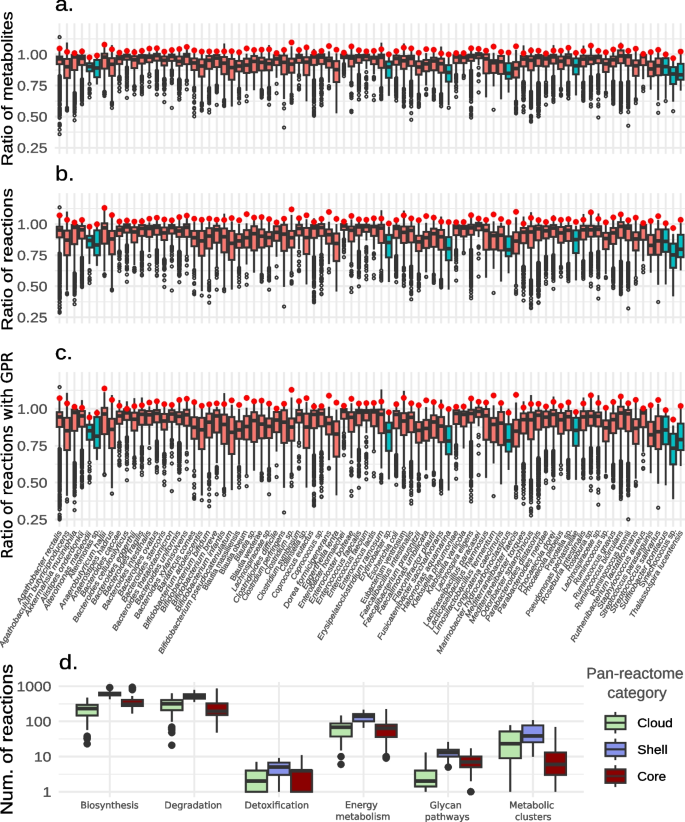
3.4 pan-GEM结构与功能质量评估
实验目的:评估pan-GEMs的结构完整性和功能预测能力,验证pan-Draft工具的性能。
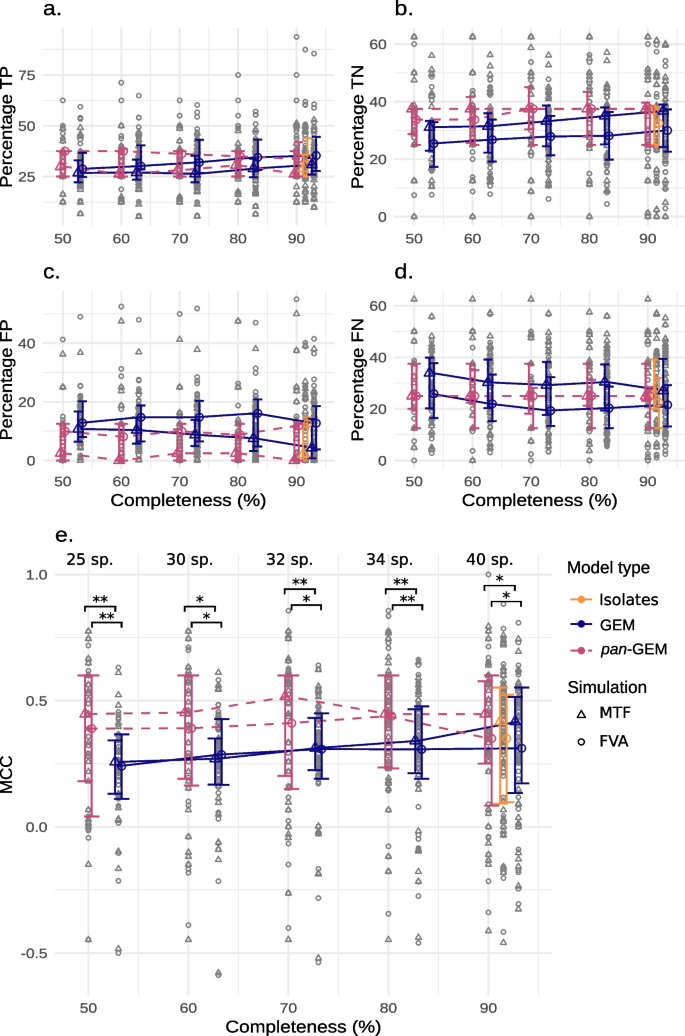
方法细节:结构评估方面,比较pan-GEMs与MAG-GEMs、iso-GEMs的代谢物和反应数量,分析代谢反应的功能分类;功能评估方面,选取41种人类肠道微生物,通过通量平衡分析(FBA)和通量变异性分析(FVA)预测厌氧发酵产物,比较pan-GEMs与MAG-GEMs、iso-GEMs及MIGRENE模型的预测准确性。
结果解读:结构评估显示,pan-GEMs的代谢物数量比iso-GEMs多3.1±2.1%,反应数量多4.0±2.7%,核心代谢反应(频率>95%)主要集中在脂质、核苷酸、辅因子的生物合成及碳水化合物降解等通路,结构完整性显著优于MAG-GEMs;功能评估显示,pan-GEMs的发酵产物预测准确性显著高于MAG-GEMs(配对t检验,P<0.05),与iso-GEMs相当,且优于MIGRENE模型,MCC值分别提高0.20(FVA)和0.18(MTF分析),能有效减少假阳性预测。
产品关联:文献未提及具体实验产品,领域常规使用sybil包进行通量分析、CPLEX求解器进行优化计算。
4. Biomarker研究及发现成果解析
本研究中的Biomarker为物种水平的核心代谢反应集合及对应的最小反应频率(MRF)阈值,其筛选逻辑为基于同一物种多个MAGs的反应频率统计,通过与参考基因组GEMs的比较确定最优阈值,实现核心代谢反应的精准筛选。
Biomarker定位:Biomarker类型为物种水平的核心代谢反应集合,筛选逻辑为:首先基于UHGG和OMD数据库的MAGs数据集,统计每个反应在同一物种MAGs中的出现频率;然后通过与参考基因组GEMs的反应组比较,确定能最大化模型准确性的MRF阈值(6%);最终筛选出频率≥6%的反应作为物种的核心代谢反应,构建pan-GEMs。
研究过程详述:Biomarker的来源为宏基因组组装基因组(MAGs)的代谢模型,验证方法为将筛选得到的核心反应组与参考基因组的反应组进行比较,计算Matthews相关系数(MCC)评估准确性;特异性与敏感性数据显示,6%的MRF阈值下,pan-GEMs与参考模型的MCC最高,能有效排除污染反应(频率<6%的反应多为污染或附属代谢反应),同时保留90%以上的核心代谢反应;当MAG数量≥30时,该阈值的特异性和稳定性最佳。
核心成果提炼:该Biomarker(核心代谢反应集合)的功能关联为能准确反映物种的真实代谢能力,作为物种水平代谢模型的核心框架,可用于预测微生物的厌氧发酵产物等功能;创新性在于首次提出基于泛反应组频率的核心代谢反应筛选策略,无需参考基因组,适用于未培养物种;统计学结果显示,与MIGRENE模型相比,pan-GEMs的发酵产物预测MCC值分别提升0.20(FVA分析)和0.18(MTF分析),与MAG-GEMs相比,预测准确性显著提高(配对t检验,P<0.05),说明该Biomarker能有效提高代谢模型的功能预测能力。