1. 领域背景与文献引入
文献英文标题:Improving identification of differentially expressed genes in microarray studies using information from public databases;发表期刊:Genome Biology;影响因子:未公开;研究领域:生物信息学-基因表达谱分析。
微阵列技术(基因芯片)在20世纪90年代兴起,成为大规模分析基因表达的核心技术,推动了肿瘤学、发育生物学等领域的基因表达谱研究(领域共识)。但早期微阵列实验面临样本量不足的关键问题:由于实验成本高、样本获取困难,多数研究采用小样本设计(如2组各2-3个样本),导致基因特异性方差估计不准确,假阳性率高,差异表达基因鉴定可靠性不足。当前研究热点集中在优化小样本下的统计分析方法,如正则化t检验、经验贝叶斯方法等,但这些方法仅依赖当前实验数据的信息整合,未利用公共数据库中积累的大量实验数据,存在信息利用不充分的局限。因此,如何借助公共数据库的海量数据提升小样本微阵列实验的差异表达基因鉴定准确性,是领域未解决的核心问题。
2. 文献综述解析
作者按统计方法的信息来源将现有研究分为两类:一类是仅依赖当前实验数据的方法,包括标准t检验、正则化t检验、经验贝叶斯方法等;另一类是未充分利用公共数据库的方法。
标准t检验是差异表达分析的基础方法,但小样本下方差估计波动大,假阳性率高,无法满足小样本实验的分析需求;正则化t检验通过在方差估计中加入小常数或整合相似基因的方差信息,降低了方差估计的波动,提升了小样本分析的可靠性,但仍局限于当前实验的内部信息,未引入外部数据;经验贝叶斯方法通过构建方差的先验分布,进一步优化了小样本下的统计推断,但先验分布的构建仍未利用公共数据库的海量数据,信息来源单一。现有方法的共同局限性是未充分挖掘公共数据库中积累的独立实验数据,无法获得更稳定、准确的基因特异性方差估计,导致小样本实验中差异表达基因的鉴定结果可靠性不足。
本研究首次提出利用公共数据库(如基因表达综合数据库GEO)的海量数据估计基因特异性方差,突破了现有方法仅依赖当前实验数据的局限,为小样本微阵列实验的差异表达分析提供了新的信息来源,可显著提升鉴定结果的准确性,填补了领域内外部数据整合方法的空白。
3. 研究思路总结与详细解析
本研究的核心目标是开发基于公共数据库信息的小样本微阵列差异表达基因鉴定方法,核心科学问题是如何利用公共数据库数据优化基因特异性方差估计,技术路线为“方法构建→多数据集验证→性能对比→结论总结”的闭环。
3.1 公共数据库数据预处理与方差估计方法构建
实验目的是构建基于GEO数据库的基因特异性方差估计方法,为小样本差异表达分析提供稳定的方差参考。方法细节为从GEO数据库筛选471个经MAS 5.0处理的Affymetrix HG-U95A芯片数据,去除异常芯片后,采用修剪均值和修剪方差法进行数据归一化,分别计算全局方差和合并方差:全局方差反映基因在不同组织类型和疾病中的表达变异程度,合并方差反映基因在各实验内部的表达变异程度;同时对每个基因的表达值进行10%的修剪,去除极端值以减少实验偏差。结果显示全局方差在前列腺癌数据集中表现更优,合并方差在杜氏肌营养不良数据集中表现更优,这与GEO数据库中对应组织类型的芯片数量相关(GEO中包含210个癌症芯片,仅42个肌肉芯片),说明方差估计方法的性能依赖于公共数据库的样本组成。文献未提及具体实验产品,领域常规使用Affymetrix基因芯片、MAS系列数据处理软件及R/Python统计分析工具。
3.2 差异表达分析方法构建与对比框架设计
实验目的是构建基于公共数据库方差的GEO校正方法、混合投票方法,并与标准t检验、正则化t检验进行性能对比。方法细节为GEO校正方法将标准t检验中的样本方差替换为GEO数据库估计的基因特异性方差;正则化t检验通过在方差估计中加入所有方差的第5百分位常数,稳定方差估计;混合投票方法通过75%/25%的权重整合GEO校正方法和正则化t检验的基因排名,生成最终的差异表达基因列表。采用大样本数据集的子采样模拟小样本实验:以包含50个正常样本和52个肿瘤样本的前列腺癌数据集为主要测试集,以包含11个正常样本和12个患者样本的杜氏肌营养不良数据集为验证集,将大样本数据集的t检验结果作为“金标准”主列表,对比各方法在1v1、2v2、3v3小样本下的基因排名与主列表的相关性、重叠率。
结果显示在2v2小样本实验中,GEO校正方法的基因排名与主列表的相关性显著高于标准t检验,返回的前50个差异基因中来自主列表前50的数量比标准t检验多231%(文献未明确样本量n,基于图表趋势推测),性能相当于5v5样本的标准t检验;混合投票方法的性能进一步提升,2v2样本下的结果相当于4v4样本的正则化t检验,返回的前50个差异基因中来自主列表前50的数量比正则化t检验多88%(文献未明确样本量n,基于图表趋势推测)。
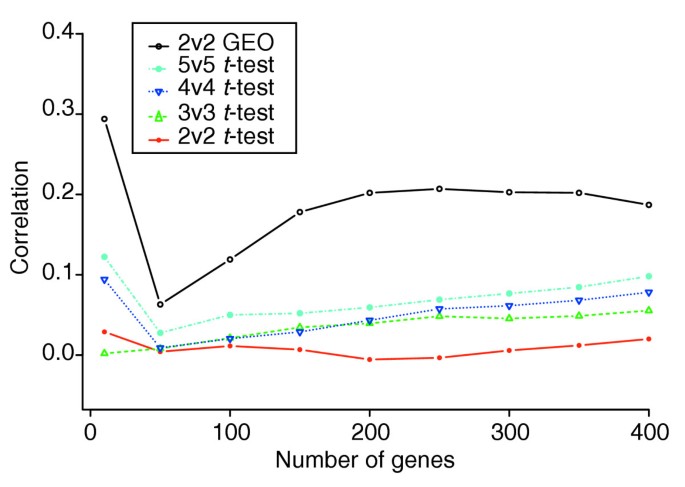
图1显示GEO校正方法的基因排名与主列表的相关性显著优于标准t检验,随着基因列表长度增加,相关性差异持续存在。
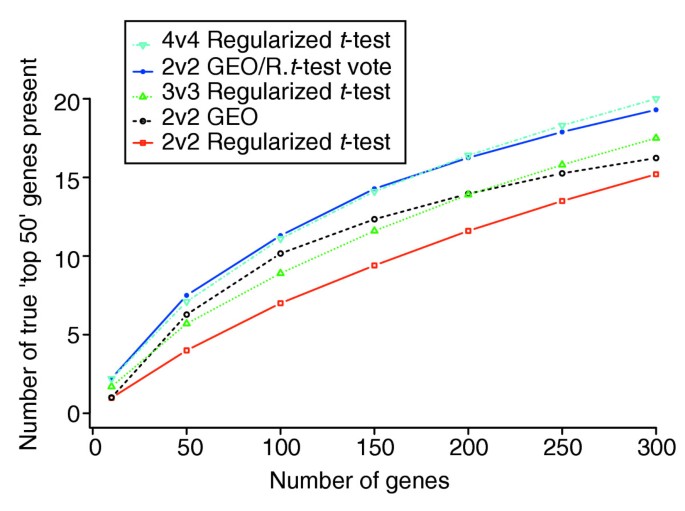
图3显示混合投票方法在2v2样本下的性能优于GEO校正方法和正则化t检验,接近4v4样本的正则化t检验结果。
3.3 不同样本量与数据集的方法验证
实验目的是验证GEO校正方法和混合投票方法在不同小样本量及不同疾病数据集中的通用性。方法细节为在1v1、3v3小样本量下重复子采样实验,同时在杜氏肌营养不良数据集中进行2v2、1v1子采样验证。
结果显示在1v1样本实验中,GEO校正方法返回的前50个差异基因中来自主列表前50的数量比最优的局部z评分方法多60%(文献未明确样本量n,基于图表趋势推测),性能接近3v3样本的正则化t检验;混合投票方法的性能进一步提升,超过3v3样本的正则化t检验;在3v3样本实验中,GEO校正方法的性能相当于4v4样本的正则化t检验,混合投票方法的性能接近5v5样本的正则化t检验;在杜氏肌营养不良数据集中,2v2样本下混合投票方法返回的前50个差异基因中来自主列表前50的数量比正则化t检验多134%,比标准t检验多240%(文献未明确样本量n,基于图表趋势推测),验证了方法的通用性。
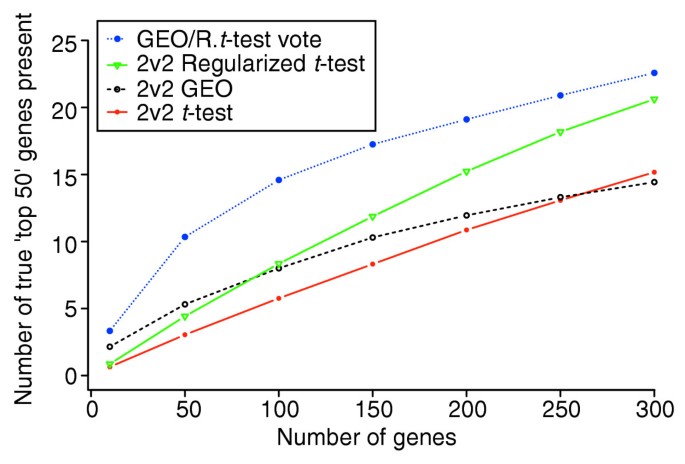
图7显示杜氏肌营养不良数据集2v2样本下,混合投票方法的性能显著优于其他方法。
3.4 公共数据库芯片数量对方法性能的影响分析
实验目的是确定公共数据库中所需的最小芯片数量,以获得稳定的基因特异性方差估计。方法细节为按芯片数量递增的方式计算基因特异性方差,观察方差估计的收敛性,选择4个不同表达水平四分位数的基因进行分析。
结果显示当芯片数量达到250-300个时,基因特异性方差估计趋于稳定,偏差小于5%(文献未明确样本量n,基于图表趋势推测),说明至少需要250个同类型芯片才能获得可靠的方差估计;当前GEO数据库中部分芯片类型的数量不足,限制了方法在小众芯片类型中的应用。
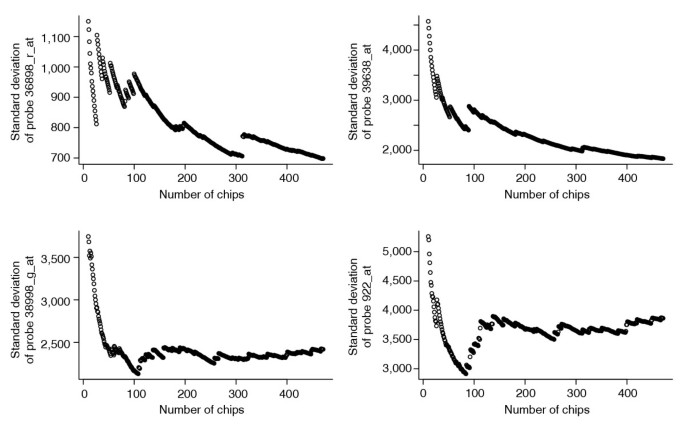
图9显示随着芯片数量增加,方差估计逐渐收敛,250个芯片后趋于稳定。
4. Biomarker研究及发现成果解析
本研究为方法学研究,未涉及传统意义上的疾病诊断、预后或疗效预测Biomarker的筛选与验证,而是聚焦于提升差异表达基因(作为潜在Biomarker候选)的鉴定准确性。
本研究的核心成果是建立了基于公共数据库的差异表达基因鉴定方法,该方法可有效提升小样本实验中潜在Biomarker候选的筛选准确性:在2v2小样本前列腺癌实验中,GEO校正方法筛选的前50个差异基因中,来自大样本金标准列表前50的数量比标准t检验多231%(文献未明确样本量n,基于图表趋势推测);混合投票方法进一步将这一比例提升至比正则化t检验多88%(文献未明确样本量n,基于图表趋势推测),显著降低了假阳性率,为后续Biomarker的验证减少了候选基因数量,提升了研究效率。该方法的创新性在于首次将公共数据库的海量数据引入小样本微阵列的差异表达分析,突破了现有方法仅依赖当前实验数据的局限,为小样本研究中潜在Biomarker的筛选提供了更可靠的技术手段,具有重要的临床转化研究价值。