1. 领域背景与文献引入
文献英文标题:LncRNA APCDD1L-AS1 induces icotinib resistance by inhibition of EGFR autophagic degradation via the miR-1322/miR-1972/miR-324-3p-SIRT5 axis in lung adenocarcinoma;发表期刊:Biomarker Research;影响因子:未公开;研究领域:肺腺癌靶向治疗耐药机制。
肺癌是全球发病率和死亡率最高的恶性肿瘤之一,肺腺癌作为非小细胞肺癌的主要亚型,约占所有病例的40%。过去十年,表皮生长因子受体酪氨酸激酶抑制剂(EGFR-TKI)的问世极大改善了携带EGFR敏感突变的肺腺癌患者的生存预后,其中埃克替尼作为我国自主研发的一代EGFR-TKI,已成为此类患者的标准一线治疗方案。然而,多数患者在接受治疗10-16个月后会出现获得性耐药,成为临床治疗的核心障碍。目前已知的耐药机制包括EGFR T790M二次突变、MET基因扩增等,针对这些机制的三代EGFR-TKI或MET抑制剂已应用于临床,但仍有超过30%的患者缺乏明确的耐药机制及有效的治疗策略,因此亟需探索新的耐药调控通路。
随着非编码RNA研究的深入,长链非编码RNA(lncRNA)已被证实参与肿瘤细胞增殖、凋亡、耐药等多种生物学过程,其通过竞争性内源性RNA(ceRNA)机制调控靶基因表达的模式成为研究热点。已有研究发现部分lncRNA参与EGFR-TKI耐药,但相关研究多聚焦于单个lncRNA与单个miRNA的相互作用,针对多miRNA协同参与的ceRNA网络调控耐药的研究仍较为匮乏。本研究针对这一空白,系统探索了lncRNA APCDD1L-AS1在肺腺癌埃克替尼耐药中的作用及分子机制,为耐药机制研究提供了新的视角。
2. 文献综述解析
作者对领域内现有研究的评述逻辑主要分为两个维度:一是EGFR-TKI耐药的已知分子机制,二是lncRNA在肿瘤耐药中的调控模式。
现有研究表明,EGFR-TKI耐药的核心机制可分为靶基因依赖和非靶基因依赖两类,靶基因依赖机制以EGFR T790M突变最为常见,约占获得性耐药的50%,三代EGFR-TKI奥希替尼可特异性抑制该突变型EGFR;非靶基因依赖机制包括MET基因扩增、PI3K-AKT-mTOR通路激活等,联合MET抑制剂或mTOR抑制剂可部分逆转耐药。在lncRNA领域,已有研究证实lncRNA可通过ceRNA机制调控肿瘤耐药,例如lncRNA MALAT1通过海绵吸附miR-200a上调ZEB1表达导致吉非替尼耐药,lncRNA GSTM3TV2通过海绵吸附let-7上调LAT2和OLR1表达促进胰腺癌吉西他滨耐药,但这些研究多聚焦于单一lncRNA-miRNA-mRNA的调控轴,缺乏对多miRNA协同参与的稳定ceRNA网络的探索,且针对肺腺癌EGFR-TKI耐药的lncRNA研究仍相对有限,具体调控机制尚未完全阐明。
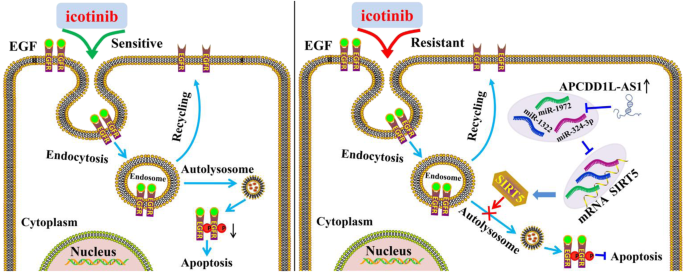
本研究的创新价值在于首次构建了lncRNA APCDD1L-AS1同时海绵吸附三个miRNA的ceRNA网络,通过解除miR-1322/miR-1972/miR-324-3p对SIRT5的转录抑制,进而抑制EGFR自噬降解促进埃克替尼耐药,弥补了现有研究中单一ceRNA调控轴功能较弱的局限性,为EGFR-TKI耐药机制研究提供了新的范式。
3. 研究思路总结与详细解析
本研究的核心目标是明确lncRNA APCDD1L-AS1在肺腺癌埃克替尼耐药中的作用及分子机制,核心科学问题是APCDD1L-AS1如何调控EGFR的稳定性以介导耐药,技术路线遵循“耐药细胞模型构建与验证→差异lncRNA筛选→功能验证→分子机制解析→体内模型验证”的闭环逻辑,通过细胞实验、分子实验、动物实验多层面验证研究假设。
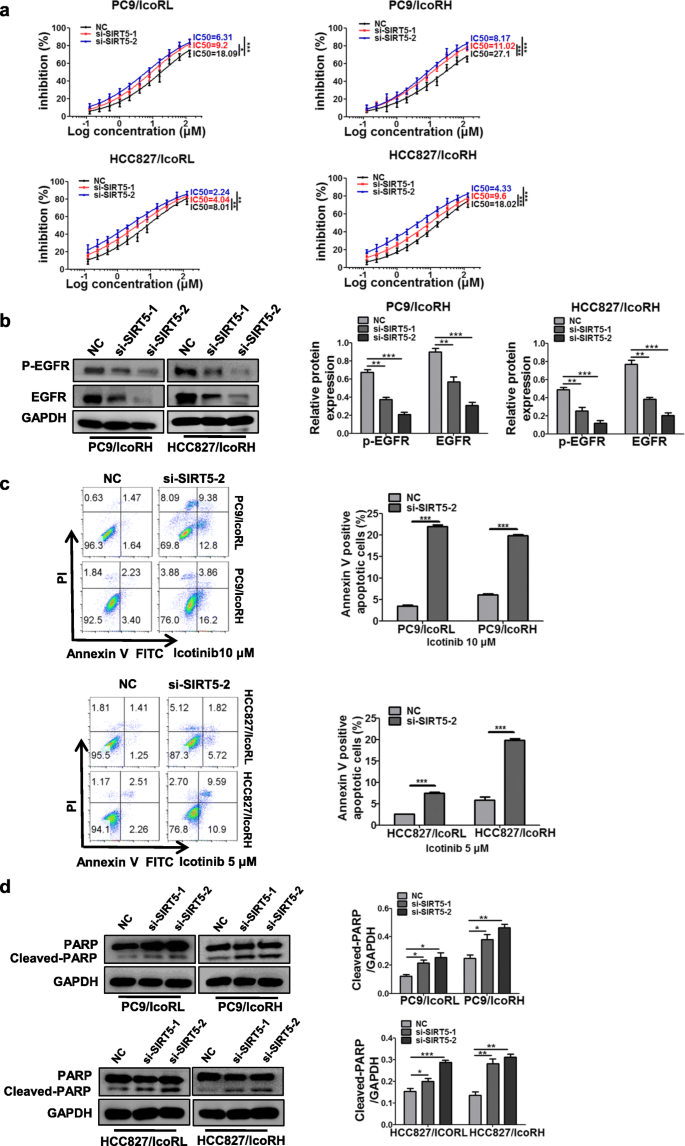
3.1 埃克替尼耐药细胞模型构建与表型验证
实验目的:构建并验证埃克替尼耐药的肺腺癌细胞模型,明确耐药细胞的表型特征及EGFR表达变化。
方法细节:采用梯度浓度埃克替尼(0.01-20μM)处理人肺腺癌细胞系PC9和HCC827超过6个月,构建低耐药(PC9/IcoRL、HCC827/IcoRL)和高耐药(PC9/IcoRH、HCC827/IcoRH)细胞系;通过MTT法检测不同浓度埃克替尼处理96小时后的细胞存活率,计算半数抑制浓度(IC50);检测10μM埃克替尼处理24、48、72、96小时后的细胞活力;通过克隆形成实验分析不同浓度埃克替尼处理后的克隆形成能力;构建裸鼠皮下移植瘤模型,给予埃克替尼处理后每周测量肿瘤体积(n=3);通过蛋白质免疫印迹(Western Blot)检测EGFR蛋白表达及磷酸化水平。
结果解读:MTT实验显示,耐药细胞的IC50值显著高于亲本细胞,且耐药性呈剂量和时间依赖性;克隆形成实验显示,埃克替尼处理后亲本细胞的克隆数量和大小显著小于耐药细胞;裸鼠移植瘤模型显示,埃克替尼处理后亲本细胞组的肿瘤体积显著小于耐药细胞组(n=3,P<0.01);蛋白质免疫印迹结果显示,耐药细胞中EGFR蛋白表达及磷酸化水平显著高于亲本细胞。以上结果证实了耐药细胞模型的成功构建,且耐药细胞中EGFR的稳定性和活性显著升高。
产品关联:实验所用关键产品:MTT(Sigma-Aldrich, USA)、EGFR抗体(Cell Signaling Technology, USA)、磷酸化EGFR抗体(Cell Signaling Technology, USA)、GAPDH抗体(Cell Signaling Technology, USA)。
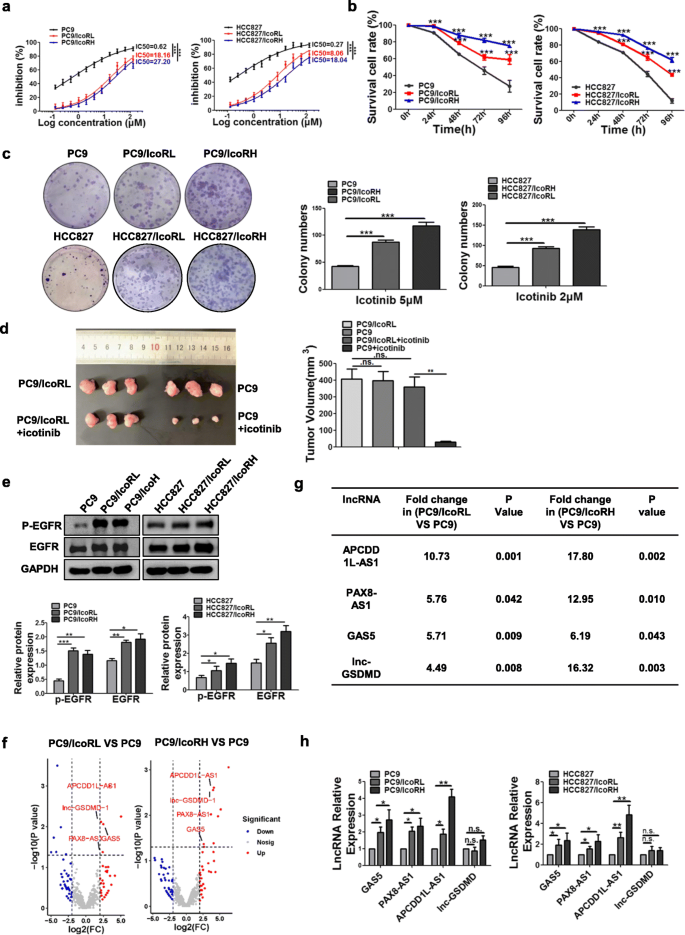
3.2 差异表达lncRNA筛选与APCDD1L-AS1的功能验证
实验目的:筛选耐药细胞中差异表达的lncRNA,并验证APCDD1L-AS1在埃克替尼耐药中的功能及临床意义。
方法细节:通过转录组测序分析PC9、PC9/IcoRL、PC9/IcoRH细胞中的lncRNA表达谱,筛选差异表达的lncRNA;通过实时荧光定量PCR(qRT-PCR)验证候选lncRNA的表达水平;通过编码潜能分析工具(CPAT)验证APCDD1L-AS1的非编码特性;通过核质分离qRT-PCR和RNA荧光原位杂交(FISH)检测APCDD1L-AS1的亚细胞定位;通过小干扰RNA(siRNA)敲低或过表达载体过表达APCDD1L-AS1,检测埃克替尼IC50值、EGFR表达及磷酸化水平;通过流式细胞术和蛋白质免疫印迹检测细胞凋亡情况;通过Kaplan-Meier Plotter数据库分析APCDD1L-AS1表达与肺腺癌患者总生存期的相关性。
结果解读:转录组测序显示,APCDD1L-AS1是耐药细胞中上调最显著的lncRNA之一,qRT-PCR验证了其在耐药细胞中的高表达(P<0.001);编码潜能分析显示APCDD1L-AS1无蛋白编码能力;核质分离和FISH实验显示APCDD1L-AS1主要定位于细胞质;敲低APCDD1L-AS1可显著降低耐药细胞的埃克替尼IC50值,增强敏感性,同时降低EGFR表达及磷酸化水平(P<0.01);Kaplan-Meier分析显示,APCDD1L-AS1高表达的肺腺癌患者总生存期显著短于低表达患者(log rank test,P=0.0048);流式细胞术和蛋白质免疫印迹显示,敲低APCDD1L-AS1可显著促进埃克替尼诱导的细胞凋亡(P<0.01)。以上结果证实APCDD1L-AS1可促进肺腺癌埃克替尼耐药,并与患者不良预后相关。
产品关联:实验所用关键产品:Trizol试剂(Invitrogen, Carlsbad, CA, USA)、PrimeScript™ RT试剂试剂盒(Takara, Tokyo, Japan)、SYBR Premix ExTaq II试剂盒(TaKaRa, Japan)、siRNA(Ribobio, Guangzhou, China)、Annexin V-FITC凋亡检测试剂盒、流式细胞仪(BD Biosciences Inc., Franklin, NJ, USA)、PARP抗体(Cell Signaling Technology, USA)。
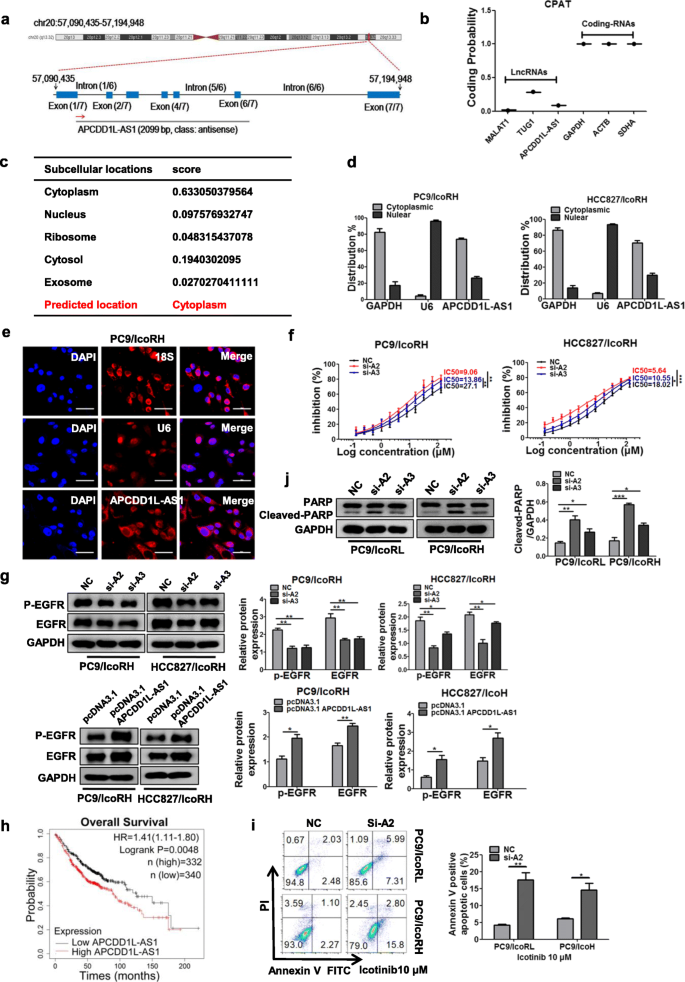
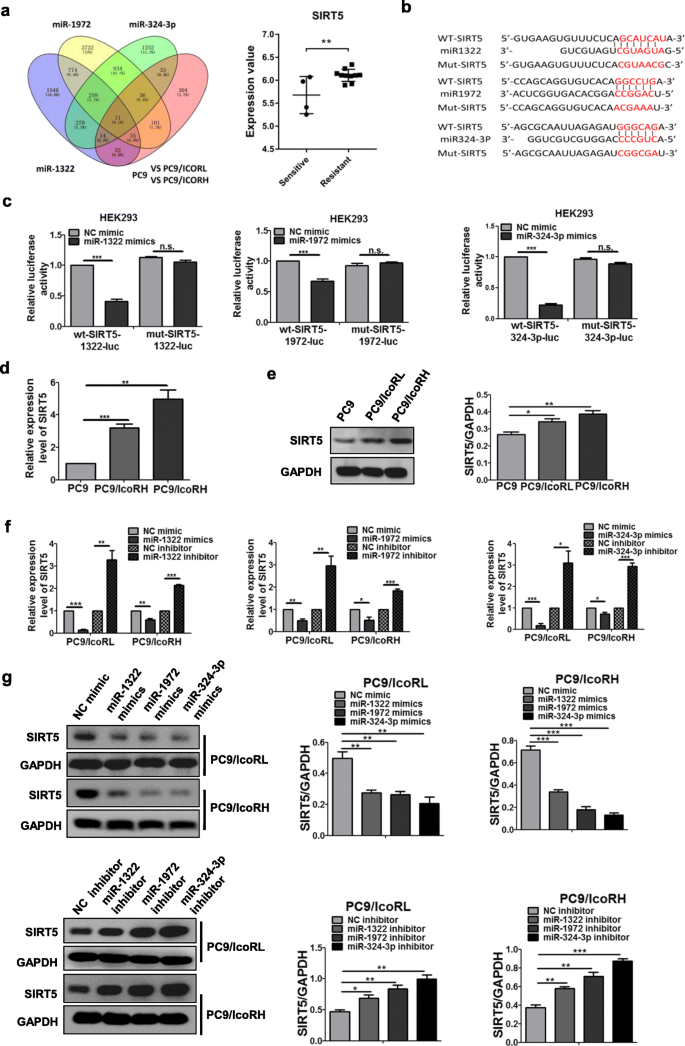
3.3 APCDD1L-AS1与miRNA的相互作用验证
实验目的:验证APCDD1L-AS1作为ceRNA海绵吸附的miRNA,并明确其在耐药中的介导作用。
方法细节:通过LncBase V2.0数据库预测与APCDD1L-AS1结合的miRNA,筛选出miR-1322、miR-1972、miR-324-3p;通过双荧光素酶报告基因实验验证APCDD1L-AS1与这些miRNA的直接结合;通过RNA免疫沉淀(RIP)实验验证APCDD1L-AS1与AGO2及这些miRNA的共沉淀;通过实时荧光定量PCR检测亲本细胞和耐药细胞中这些miRNA的表达水平;通过转染miRNA模拟物或抑制剂,检测埃克替尼敏感性、EGFR表达及磷酸化水平、细胞凋亡情况;通过共转染siRNA-APCDD1L-AS1和miRNA抑制剂,验证miRNA在APCDD1L-AS1调控耐药中的介导作用。
结果解读:双荧光素酶报告基因实验显示,miRNA模拟物可显著抑制野生型APCDD1L-AS1报告载体的荧光素酶活性,对突变型载体无影响(P<0.01);RIP实验显示,APCDD1L-AS1与miR-1322、miR-1972、miR-324-3p均可与AGO2共沉淀(P<0.05);实时荧光定量PCR显示,耐药细胞中这三个miRNA的表达水平显著低于亲本细胞(P<0.05);转染miRNA模拟物可显著降低耐药细胞的埃克替尼IC50值,增强敏感性,同时降低EGFR表达及磷酸化水平,促进细胞凋亡(P<0.01);共转染siRNA-APCDD1L-AS1和miRNA抑制剂可部分逆转敲低APCDD1L-AS1导致的敏感性增强(P<0.05)。以上结果证实APCDD1L-AS1通过海绵吸附miR-1322、miR-1972、miR-324-3p介导埃克替尼耐药。
产品关联:实验所用关键产品:双荧光素酶报告基因检测试剂盒(Promega, Madison, WI, USA)、Magna RNA结合蛋白免疫沉淀试剂盒(Millipore, Billerica, MA, USA)、AGO2抗体(Abcam, Cambridge, USA)、miRNA模拟物/抑制剂(Ribobio, Guangzhou, China)。
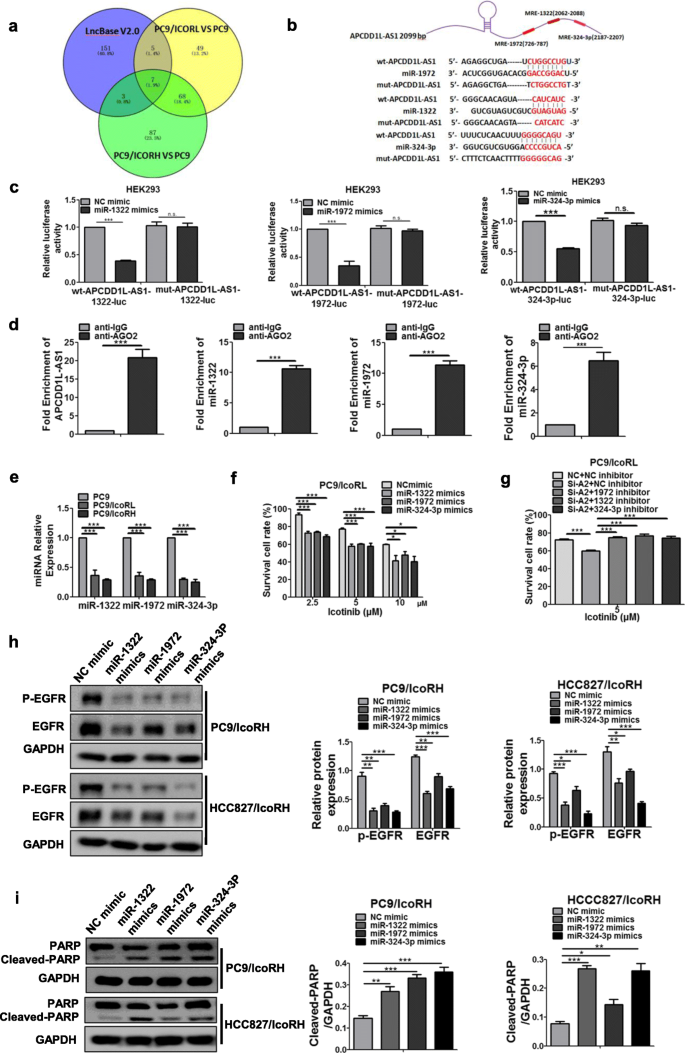
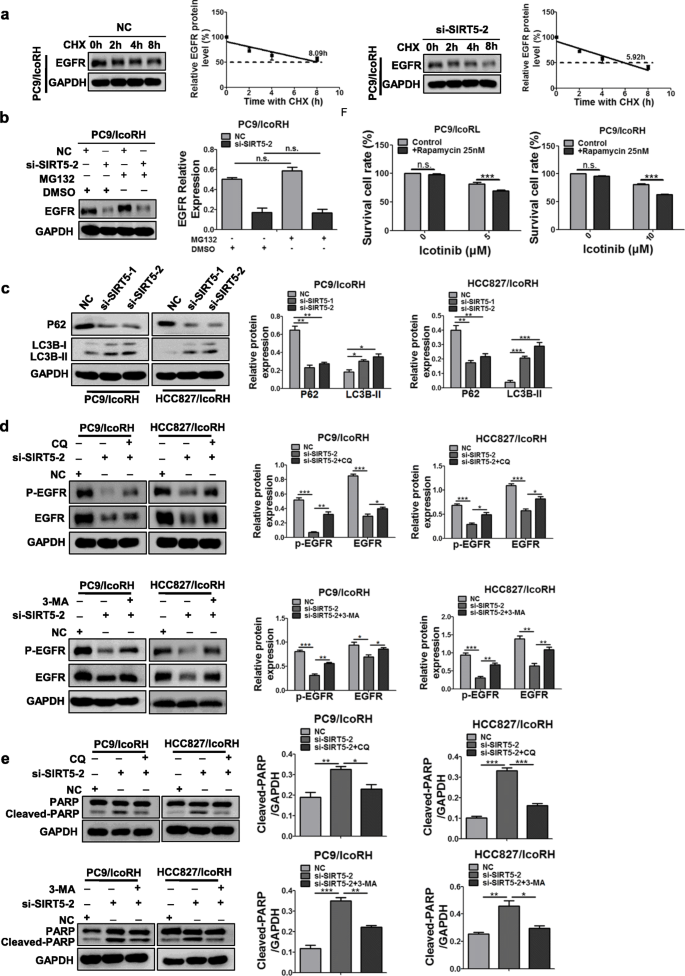
3.4 miRNA下游靶基因SIRT5的验证及功能分析
实验目的:明确miRNA的共同靶基因,并验证其在耐药中的作用及调控EGFR的机制。
方法细节:通过Targetscan数据库预测和转录组测序差异基因分析,筛选三个miRNA的共同靶基因SIRT5;通过双荧光素酶报告基因实验验证miRNA与SIRT5 3"UTR的直接结合;通过实时荧光定量PCR和蛋白质免疫印迹检测亲本细胞和耐药细胞中SIRT5的表达水平;通过转染miRNA模拟物或抑制剂,检测SIRT5的表达变化;通过siRNA敲低SIRT5,检测埃克替尼IC50值、EGFR表达及磷酸化水平、细胞凋亡情况;通过蛋白质合成抑制剂环己酰亚胺(CHX)处理,检测EGFR蛋白半衰期;通过自噬抑制剂氯喹(CQ)、3-甲基腺嘌呤(3-MA)处理,验证自噬在EGFR降解中的作用;通过共聚焦显微镜观察EGFR与自噬标记物LC3B的共定位。
结果解读:双荧光素酶报告基因实验显示,miRNA模拟物可显著抑制野生型SIRT5 3"UTR报告载体的荧光素酶活性(P<0.01);实时荧光定量PCR和蛋白质免疫印迹显示,耐药细胞中SIRT5的表达水平显著高于亲本细胞(P<0.001);转染miRNA模拟物可降低SIRT5表达,抑制剂则升高其表达(P<0.05);敲低SIRT5可显著降低耐药细胞的埃克替尼IC50值,降低EGFR表达及磷酸化水平,促进细胞凋亡(P<0.01);CHX处理显示,敲低SIRT5可显著缩短EGFR的蛋白半衰期(P<0.05);自噬抑制剂可部分逆转敲低SIRT5导致的EGFR下调(P<0.05);共聚焦显微镜显示,敲低SIRT5后EGFR与LC3B的共定位增加。以上结果证实SIRT5是三个miRNA的共同靶基因,通过抑制EGFR自噬降解促进埃克替尼耐药。
产品关联:实验所用关键产品:CHX(Sigma-Aldrich, USA)、CQ(Sigma-Aldrich, St. Louis, MO, USA)、3-MA(Selleck Chemicals, Houston, TX, USA)、SIRT5抗体(Sigma-Aldrich, USA)、LC3B抗体(Cell Signaling Technology, USA)、p62抗体(Cell Signaling Technology, USA)。
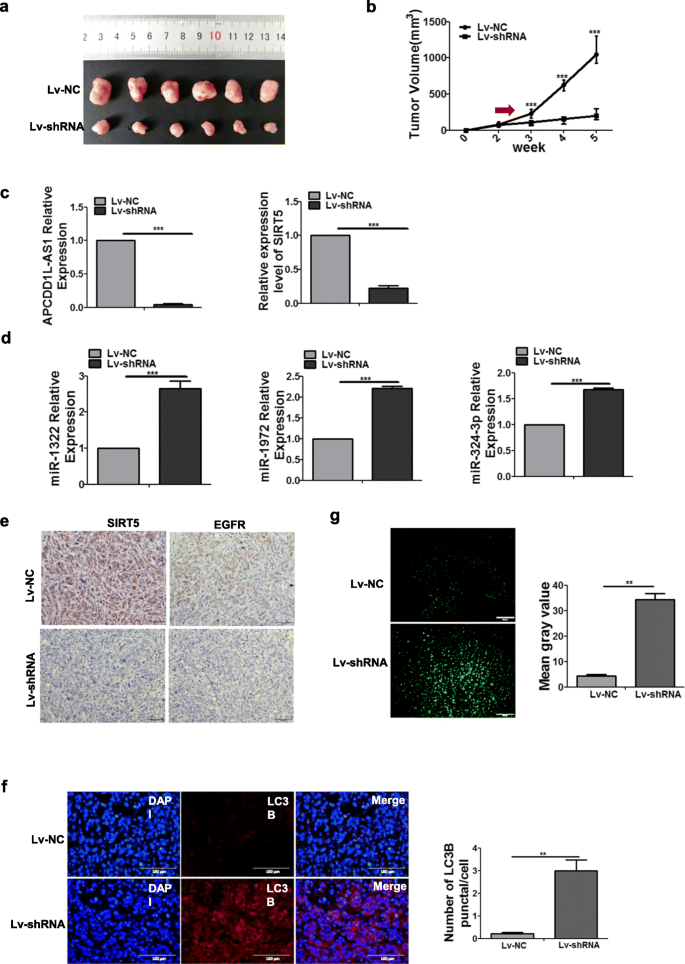
3.5 体内模型验证APCDD1L-AS1的调控机制
实验目的:在体内水平验证APCDD1L-AS1介导埃克替尼耐药的分子机制。
方法细节:构建稳定敲低APCDD1L-AS1的PC9/IcoRH细胞系(Lv-shRNA-APCDD1L-AS1),并构建裸鼠皮下移植瘤模型(n=6);给予埃克替尼处理后每周测量肿瘤体积;通过实时荧光定量PCR检测肿瘤组织中APCDD1L-AS1、SIRT5及miRNA的表达水平;通过免疫组化(IHC)检测肿瘤组织中SIRT5和EGFR的表达;通过免疫荧光检测LC3B puncta的数量;通过TUNEL实验检测细胞凋亡情况。
结果解读:埃克替尼处理后,Lv-shRNA-APCDD1L-AS1组的肿瘤体积显著小于对照组(n=6,P<0.01);实时荧光定量PCR显示,敲低组中APCDD1L-AS1和SIRT5的表达显著降低,miR-1322、miR-1972、miR-324-3p的表达显著升高(P<0.001);免疫组化显示,敲低组中SIRT5和EGFR的表达显著降低;免疫荧光显示,敲低组中LC3B puncta数量显著增加(P<0.01);TUNEL实验显示,敲低组的细胞凋亡水平显著高于对照组(Lv-NC组平均灰度值为4.39±0.568,Lv-shRNA组为34.39±2.273,P<0.001)。以上结果在体内水平证实了APCDD1L-AS1通过miR-1322/miR-1972/miR-324-3p-SIRT5轴抑制EGFR自噬降解,促进埃克替尼耐药。
产品关联:实验所用关键产品:慢病毒载体(OBiotech, Shanghai, China)、嘌呤霉素(Sigma-Aldrich, USA)、免疫组化抗体SIRT5(Sigma-Aldrich, USA)、EGFR(Santa, Clara, CA, USA)、TUNEL凋亡检测试剂盒(Byotime, Jiangsu, China)。
4. Biomarker研究及发现成果解析
本研究鉴定了以lncRNA APCDD1L-AS1为核心的耐药相关Biomarker网络,包括miR-1322、miR-1972、miR-324-3p及SIRT5,为肺腺癌EGFR-TKI耐药的诊断和治疗提供了新的靶点。
Biomarker定位:APCDD1L-AS1作为核心耐药Biomarker,筛选逻辑为“转录组测序筛选耐药细胞中差异上调lncRNA→细胞系qRT-PCR验证→临床预后相关性分析”;miR-1322、miR-1972、miR-324-3p作为APCDD1L-AS1的下游调控分子,通过生物信息学预测和双荧光素酶报告基因实验验证;SIRT5作为miRNA的共同靶基因,通过靶基因预测和功能实验验证,形成了完整的“lncRNA-miRNA-mRNA”调控轴。
研究过程详述:APCDD1L-AS1来源于肺腺癌细胞系的转录组,通过实时荧光定量PCR在亲本细胞和耐药细胞中验证其表达,耐药细胞中表达水平显著高于亲本细胞(P<0.001);通过Kaplan-Meier Plotter数据库分析672例肺腺癌患者的临床数据,显示APCDD1L-AS1高表达患者的总生存期显著短于低表达患者(log rank test,P=0.0048)。miR-1322、miR-1972、miR-324-3p在耐药细胞中表达显著下调(P<0.05),转染模拟物可显著增强埃克替尼敏感性(IC50降低,P<0.01)。SIRT5在耐药细胞中表达显著上调(P<0.001),敲低SIRT5可显著降低埃克替尼IC50值(P<0.01)。
核心成果提炼:APCDD1L-AS1作为新型耐药Biomarker,可独立预测肺腺癌患者的不良预后(P=0.0048);其通过调控miR-1322/miR-1972/miR-324-3p-SIRT5轴抑制EGFR自噬降解,首次揭示了lncRNA介导的多miRNA协同调控EGFR稳定性的耐药机制;此外,自噬诱导剂雷帕霉素与埃克替尼联合可部分逆转耐药,为临床克服EGFR-TKI耐药提供了新的策略。