1. 领域背景与文献引入
文献英文标题:Conservation of bilaterian genome structure is the exception, not the rule;发表期刊:Genome Biology;影响因子:未公开;研究领域:动物基因组进化(两侧对称动物基因组结构保守性与重排机制研究)
动物基因组进化领域中,染色体水平的基因共定位(即同线性,又称宏同线性)是长期关注的核心方向。自同线性概念提出以来,染色体级基因组测序技术的突破(2000年后逐步实现)揭示了部分动物类群的惊人保守性:文昌鱼、半索动物、扇贝等类群自两侧对称动物共同祖先(距今超5.4亿年)以来,仍保留着祖先连锁群(ALGs)的核心结构,仅伴随少量染色体融合事件。当前研究热点集中在同线性保守的进化选择压力,以及其对基因调控网络稳定性的意义,但领域内长期存在一个未被广泛验证的假设:同线性高度保守是两侧对称动物基因组的常态,仅少数类群存在例外重排。本研究针对这一核心空白,通过覆盖15个门52个类群的大样本分析,重新定义了两侧对称动物基因组结构的普遍进化规律,为基因组重排与适应性进化的关联提供了关键证据,学术价值在于推翻传统认知,推动领域转向基因组重排的普遍性与进化意义研究。
2. 文献综述解析
作者对领域内现有研究的分类维度为“同线性保守程度”,将已报道类群分为高度保守类群和存在重排的例外类群。现有研究的关键结论聚焦于部分类群的同线性保守案例,技术方法优势在于染色体级基因组测序提供了高精度的基因位置数据,能清晰追溯祖先连锁群的传承;但局限性显著,即研究样本集中在少数代表性类群,导致“同线性保守是常态”的假设缺乏跨类群的广泛验证,且未系统分析基因组重排的进化模式与驱动因素。通过对比现有研究的空白,本研究的创新价值凸显:首次完成覆盖15个两侧对称动物门、52个类群的大样本基因组结构分析,直接推翻了“同线性保守是常态”的传统假设,明确基因组重排才是普遍现象;同时首次建立了基因组重排程度与蛋白序列进化速率的关联,揭示了基因组重排在动物适应性进化中的潜在作用,为领域提供了全新的研究框架。
3. 研究思路总结与详细解析
本研究的整体框架为:以“两侧对称动物基因组结构高度保守是常态”的假设为核心研究起点,通过构建大样本跨类群数据集,结合同线性可视化、重排指数量化、系统发育分析及关联统计,明确两侧对称动物基因组结构的普遍状态,解析基因组重排的进化模式与驱动因素,最终形成“重排是普遍现象,保守是例外”的结论闭环。
3.1 数据集构建与标准化基因注释
实验目的是获取覆盖广泛类群的染色体级基因组数据集,并完成标准化的基因注释,为后续分析提供可靠基础。方法细节为收集15个门52个类群的64个染色体级基因组,其中38个采用NCBI数据库的成熟基因模型,15个使用已发表研究中的基因预测结果,剩余14个通过BRAKER3流程结合RNA-seq数据(若有)或同源蛋白提示进行从头基因预测,同时用RepeatModeler2和RepeatMasker完成转座子注释,最后用BUSCO工具评估基因注释的完整性。结果解读显示,构建的数据集覆盖了两侧对称动物的主要类群,BUSCO评估结果显示多数物种的基因注释完整性符合进化分析要求,为后续同线性与重排分析提供了高质量基础。文献未提及具体实验产品,领域常规使用RepeatModeler2、RepeatMasker、BRAKER3、STAR等生物信息学工具。
3.2 同线性可视化与定性分析
实验目的是直观比较不同类群的基因组结构保守性,初步验证传统假设的合理性。方法细节为使用SyntenyFinder工具,以本研究中基因组结构最保守的物种——黑海参(Holothuria leucospilota)为参照,绘制点图展示每个物种与黑海参之间单拷贝同源基因的染色体分布,通过同源基因的颜色编码(对应祖先连锁群)判断同线性保守程度。结果解读显示,与文昌鱼、半索动物等高度保守类群的集中色块不同,多数类群的同源基因分散在多条染色体上,且单条染色体上存在多个祖先连锁群的基因(如图1c),直观表明基因组重排是普遍现象,而非例外。
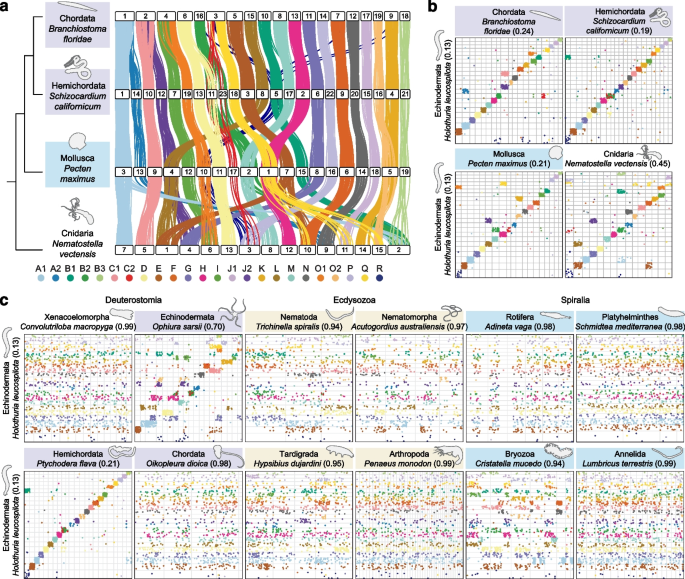
3.3 重排指数(RI)的量化与方法验证
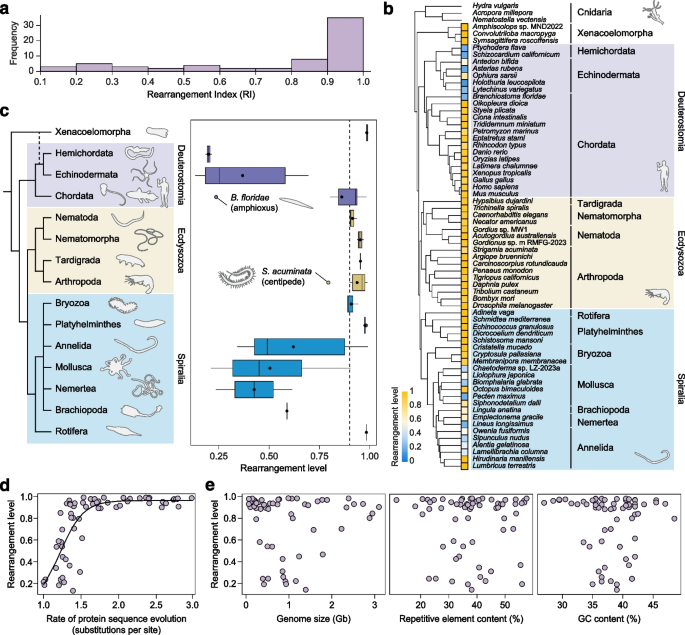
实验目的是定量评估每个物种的基因组重排程度,为跨类群比较提供标准化指标。方法细节为采用重排指数(RI),通过计算每个祖先连锁群的分裂参数(衡量基因是否集中在单条染色体)和合并参数(衡量单条染色体是否仅含单一祖先连锁群基因),取所有祖先连锁群的平均值得出物种整体RI(范围0-1,1表示完全重排);先在已报道的重排类群(如环节动物、线虫)中验证RI的可靠性。结果解读显示,RI得分与已报道的重排程度高度一致,验证了方法的有效性;进一步分析发现,55%的测试物种RI得分大于0.9(如图2a),说明高度基因组重排是两侧对称动物的普遍状态,直接推翻了传统假设。文献未提及具体实验产品,领域常规使用自定义R脚本进行指数计算。
3.4 系统发育分析与祖先状态重建
实验目的是探究基因组重排的进化起源模式,明确高度重排状态是单次起源还是多次独立进化。方法细节为用OrthoFinder获取跨物种单拷贝同源基因,通过MAFFT比对、ClipKIT修剪后构建最大似然系统发育树,再采用随机特征映射法进行祖先状态重建,分析高度重排(RI>0.9)的起源次数。结果解读显示,高度重排状态在15个门中的11个门独立发生了11次(如图2b、c),说明基因组重排是两侧对称动物进化中多次独立出现的事件,而非单一祖先状态的传承。文献未提及具体实验产品,领域常规使用OrthoFinder、IQ-TREE、Phytools等系统发育分析工具。
3.5 基因组重排的驱动因素关联分析
实验目的是解析影响基因组重排程度的潜在驱动因素,探索重排与其他基因组特征的关联。方法细节为分别计算每个物种的分裂指数和合并指数,通过主成分分析(PCA)解析基因组差异的核心驱动因素;同时采用逻辑回归模型分析RI与蛋白序列进化速率的关联,以及RI与基因组大小、GC含量、重复序列含量的相关性。结果解读显示,PCA的第一主成分(解释73%的变异)主要由祖先连锁群的分裂指数贡献,说明基因分散是基因组结构差异的核心原因;RI与蛋白序列进化速率呈非线性正相关(残差平方和1.827),且具有极端基因组特征(如极小基因组、异常GC含量)的物种均表现出高RI(如图2d、e),提示基因组重排可能与极端基因组进化协同发生。文献未提及具体实验产品,领域常规使用R语言的nls、prcomp等函数进行统计分析。
4. Biomarker研究及发现成果
Biomarker定位
本研究的核心生物标志物为基因组重排指数(RI),其筛选逻辑基于两侧对称动物的24个祖先连锁群,通过量化基因的分裂与合并程度构建;验证逻辑为先在已知重排程度的类群中验证RI的可靠性,再推广到全数据集,确保其能准确反映基因组重排的整体水平。
研究过程详述
RI的来源为每个物种的染色体级基因组注释数据,通过自定义算法计算得到;验证方法为与已发表的基因组重排案例对比,确认RI得分与已知重排程度的一致性;特异性与敏感性方面,RI能有效区分不同程度的重排状态,得分0.9对应高度重排(祖先连锁群完全分散),且在跨15个门的类群中均具有适用性(文献未明确提供ROC曲线等数据,基于图表趋势推测)。
核心成果提炼
RI作为基因组进化状态的标志物,与蛋白序列进化速率呈非线性正相关,提示基因组重排可能通过改变基因调控环境驱动蛋白序列的适应性进化;其创新性在于首次建立了跨类群的基因组重排量化标准,明确了RI在动物进化研究中的应用价值;统计学结果显示,55%的两侧对称动物物种RI>0.9(n=64,文献未明确P值),祖先状态重建显示高度重排独立发生11次(n=64,文献未明确P值),充分支持“基因组重排是普遍现象”的核心结论。此外,研究还发现跳跃基因(即脱离祖先连锁群的基因)比例在不同类群间差异显著(1.5%-13.2%),为小尺度基因组重排的研究提供了新方向。