1. 领域背景与文献引入
文献英文标题:Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies;发表期刊:Genome Biology;影响因子:17.906(2023年);研究领域:基因组学、表达数量性状位点(eQTL)与全基因组关联研究(GWAS)交叉领域。
全基因组关联研究(GWAS)自2005年首次成功应用以来,已鉴定出数千个与人类疾病和复杂表型相关的单核苷酸多态性(SNP),但超过90%的疾病关联SNP位于非编码区,其调控基因表达、影响疾病发生的分子机制仍不明确,成为领域内核心未解问题。表达数量性状位点(eQTL)分析通过关联基因型与基因表达水平,可搭建起GWAS关联SNP与功能基因之间的桥梁,是解析非编码区SNP功能的关键技术。然而早期eQTL研究存在样本量偏小(单队列样本量多不足2000人)、统计效力有限、对反式表达数量性状位点(trans-eQTL)的覆盖不足等问题,限制了对基因调控网络的全面解析。针对这一研究空白,本研究依托Framingham心脏研究的大样本队列,开展了当时规模最大的单队列全基因组eQTL分析,旨在通过大样本量提升eQTL检测的敏感性,尤其是trans-eQTL的检测,进而为GWAS关联位点的功能解读提供更全面的资源。
2. 文献综述解析
作者对领域内现有研究的分类维度主要包括样本规模、技术平台及研究类型(顺式/反式eQTL)。现有研究的关键结论显示,顺式表达数量性状位点(cis-eQTL)因调控效应更强、更易检测,已被广泛证实可有效关联GWAS SNP与邻近基因的表达,而trans-eQTL由于效应较弱,需要更大样本量才能稳定检测;技术方法上,早期研究多采用微阵列技术检测基因表达,部分研究通过meta分析整合多个队列数据提升样本量,近年出现的RNA测序技术则提供了更高分辨率的转录组信息;但现有研究存在明显局限性,如单队列样本量普遍不足2000人导致trans-eQTL检测效力不足,不同研究的技术平台差异较大,结果难以整合,且对探针多态性(polymorphism-in-probe)等实验 artifact的控制不够充分,部分研究未开展全基因组范围的trans-eQTL分析。
通过对比现有研究的未解决问题,本研究的创新价值凸显:首次采用单队列5257人的超大样本量开展全基因组eQTL分析,样本量超过此前单队列研究的2倍,大幅提升了trans-eQTL的检测效力;同时结合外显子水平的表达数据,有效识别并排除了探针多态性导致的假阳性结果;此外,本研究系统分析了trans-eQTL的聚类特征及其与GWAS表型的关联,构建了可公开访问的eQTL数据库,为GWAS关联位点的功能解读提供了更全面、可靠的资源。
3. 研究思路总结与详细解析
本研究的整体目标是构建基于大样本全血转录组的全基因组cis/trans-eQTL数据库,解析eQTL的调控特征及其与GWAS关联表型的分子机制;核心科学问题包括超大样本量能否显著提升eQTL尤其是trans-eQTL的检测能力,以及这些eQTL如何为GWAS关联位点提供功能注释;技术路线遵循“样本与数据收集→eQTL检测与质量控制→结果验证与功能分析→GWAS关联与数据库构建”的闭环逻辑,确保研究结果的可靠性与实用性。
3.1 研究样本与多组学数据收集
实验目的是获取超大样本量的全血基因表达、基因型及临床表型数据,为eQTL分析提供基础。方法细节上,研究样本来自Framingham心脏研究的两个队列:Offspring队列(2770人,2005-2008年随访)和Third Generation队列(3341人,2006-2009年随访),最终纳入5257名具有完整基因表达、基因型数据的受试者;采用PAXgene™血液RNA采集管收集空腹外周全血,室温孵育4小时稳定RNA后-80℃保存,由Asuragen公司自动化提取总RNA;基因表达检测采用Affymetrix Human Exon 1.0 ST微阵列平台,可同时检测转录簇及单个外显子的表达水平;基因型检测采用Affymetrix 500K和MIPS 50K平台,随后以1000 Genomes项目的“cosmopolitan”SNP集为参考,通过Minimac软件进行基因型填充,最终得到8510936个符合质量标准(填充质量R²≥0.3、次要等位基因频率MAF≥0.01)的SNP用于后续分析。结果解读显示,本研究的样本量为当时单队列eQTL研究之最,覆盖了更全面的基因组变异与转录组信息,为高灵敏度的eQTL检测提供了保障。实验所用关键产品:PAXgene™血液RNA采集管(PreAnalytiX)、Affymetrix Human Exon 1.0 ST微阵列、Affymetrix 500K和MIPS 50K基因分型平台。
3.2 eQTL检测与质量控制
实验目的是系统检测全基因组范围内的cis/trans-eQTL,控制混杂因素并排除实验 artifact。方法细节上,首先采用混合效应模型校正性别、年龄、血细胞计数等已知混杂因素,并通过PEER算法推断20个隐藏混杂因素,以消除群体结构、批次效应等对基因表达的影响;随后采用GPU加速的线性模型分析所有SNP-转录簇对的关联,定义cis-eQTL为位于转录起始位点(TSS)附近连续无1Mb间隙区域内的SNP,trans-eQTL为位于其他染色体或cis区域外的SNP;以错误发现率(FDR)<5%筛选显著eQTL,并通过外显子水平表达数据识别探针多态性导致的假阳性结果:当SNP仅影响单个外显子的表达关联,且基因水平关联的R²接近外显子水平时,判定为artifact。结果解读显示,本研究共检测到超过19000个独立的lead cis-eQTL和6000个独立的lead trans-eQTL,靶向超过10000个基因(eGenes),FDR<5%;其中48%的已发表GWAS显著SNP在本研究中被鉴定为显著eQTL;还发现了最长可达10Mb的长距离cis-eQTL区域,主要集中在6号染色体HLA区域等基因组连锁不平衡(LD)较强的区域;探针多态性 artifact仅影响约9.5%的eGenes,且可通过外显子水平分析有效识别。
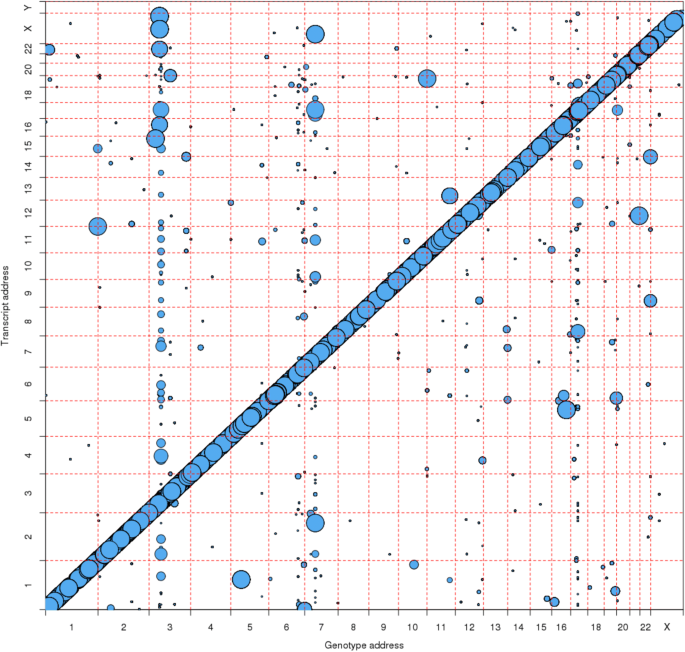
3.3 结果验证与功能富集分析
实验目的是验证eQTL结果的可靠性,并解析其调控特征与功能意义。方法细节上,内部验证将样本随机分为两个亚队列,重复eQTL分析并计算重复率;外部验证对比Westra等、Liang等、Battle等及Kirsten等已发表的eQTL研究结果,计算重复率与验证率;通过分析lead eQTL在基因结构中的位置分布,鉴定其调控偏好;采用改进的K近邻算法对trans-eQTL进行聚类分析,通过基因集富集分析(GSEA)解析聚类靶基因的功能;结合miRNA表达数据,分析miRNA在trans-eQTL调控中的作用。结果解读显示,内部验证中cis-eQTL的重复率为75%,trans-eQTL为41%,且所有重复对的表达变化方向一致;外部验证重复了Westra等研究中69%的cis-eQTL和62%的trans-eQTL,验证率显著高于随机水平;lead eQTL在基因转录区尤其是5’非翻译区(UTR)和第一外显子高度富集,提示这些区域是关键的转录调控位点;共鉴定出59个trans-eQTL聚类,每个聚类靶向6-229个trans-eGenes,其中10个聚类的靶基因显著富集miRNA靶标(FDR<5%),多个聚类与GWAS多表型关联;miRNA-mRNA共表达分析显示,部分trans-eQTL聚类的靶基因与特定miRNA表达显著相关,提示miRNA可能参与trans-eQTL的调控机制。
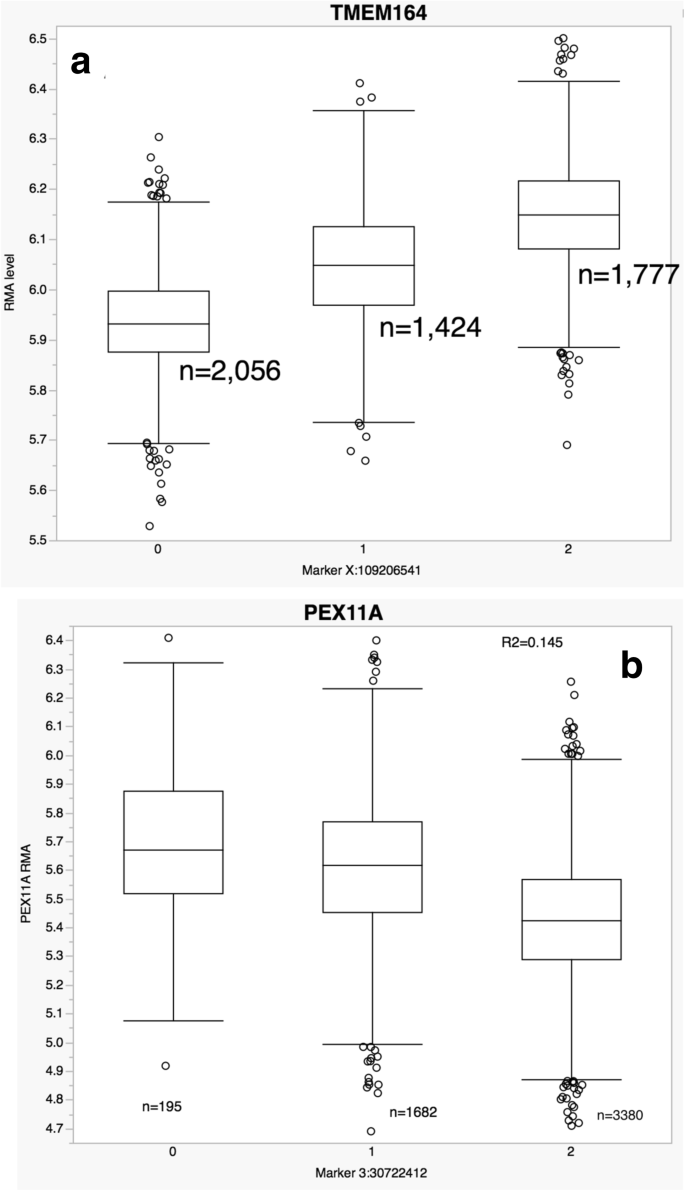
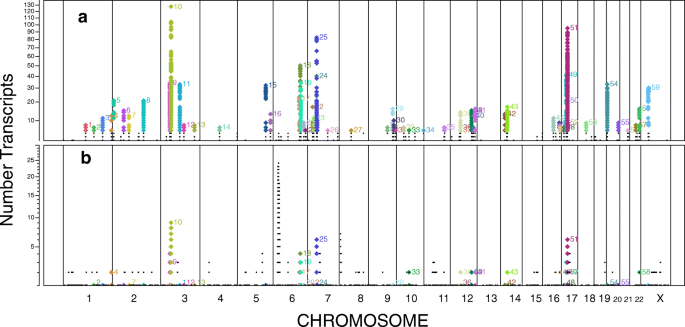
3.4 GWAS关联与数据库构建
实验目的是解析eQTL与GWAS关联表型的关联,构建可公开访问的eQTL数据库。方法细节上,将本研究的显著eQTL与NHGRI-EBI GWAS Catalog中P<5E-8的SNP进行比对,计算重叠比例;针对冠心病(CAD/MI)GWAS meta-analysis鉴定的58个位点,分析其对应的eQTL靶基因;构建NCBI Molecular QTL Browser数据库,将eQTL结果与其他研究资源整合,支持用户自定义筛选与查询。结果解读显示,48%的GWAS显著SNP在本研究中为显著eQTL,是随机预期的2倍;在58个CAD/MI GWAS位点中,21个位点存在cis-eQTL靶向34个基因,4个位点存在trans-eQTL靶向24个基因,例如LIPA基因的cis-eQTL可通过影响胆固醇酯水解参与CAD发病机制,SH2B3位点的trans-eQTL可调控干扰素信号通路基因表达;构建的数据库可通过https://preview.ncbi.nlm.nih.gov/gap/eqtl/studies/访问,为全球研究者提供eQTL查询与分析工具。
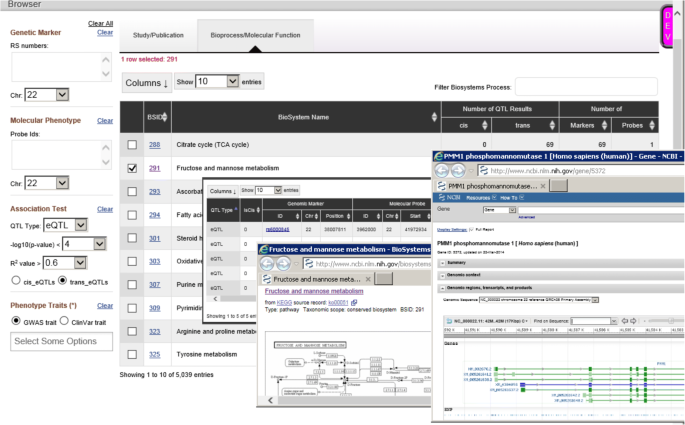
4. Biomarker研究及发现成果
本研究中的Biomarker为与GWAS关联的eQTL,包括cis-eQTL和trans-eQTL,作为连接基因型与表型的功能Biomarker。Biomarker定位上,cis-eQTL主要靶向邻近基因的表达,trans-eQTL靶向远端或其他染色体的基因表达;筛选逻辑为通过大样本全基因组关联分析,以FDR<5%筛选显著eQTL,随后与GWAS Catalog中的疾病关联SNP比对,鉴定具有临床表型关联的eQTL;验证逻辑包括内部队列重复验证、外部研究交叉验证,以及功能富集分析验证其调控合理性。
研究过程详述:Biomarker来源为5257名受试者的全血基因表达与基因型数据;验证方法包括内部队列重复(cis-eQTL重复率75%,trans-eQTL41%)、外部研究交叉验证(重复率最高达69%);特异性方面,cis-eQTL的效应值R²最高可达57%,trans-eQTL最高可达22%,显著高于随机关联;敏感性方面,大样本量使本研究检测到的cis-eGenes比单一半样本队列多60%,trans-eGenes多3-5倍。
核心成果提炼:本研究鉴定出超过19000个cis-eQTL和6000个trans-eQTL,其中48%的GWAS显著SNP为eQTL,13%为cis-eQTL,3%为trans-eQTL(n=5257,FDR<5%);在CAD/MI GWAS位点中,首次鉴定出LIPA、NT5C2等多个新的功能靶基因,例如LIPA基因的cis-eQTL rs1412445与基因表达关联的P<1E-455,可通过影响胆固醇代谢参与CAD发病;部分trans-eQTL聚类可靶向特定细胞类型的基因表达,例如血小板特异性基因、网织红细胞特异性基因,为GWAS表型提供了细胞水平的机制解释;本研究构建的eQTL数据库为GWAS关联位点的功能解读提供了重要资源,可助力疾病发病机制研究与新治疗靶点发现。