1. 领域背景与文献引入
文献英文标题:Evidence-ranked motif identification;发表期刊:Genome Biology;影响因子:未公开;研究领域:计算生物学-转录调控元件识别
随着真核生物基因组和转录组测序规模的持续扩大,解析复杂生物体内基因间的功能调控关系成为生命科学领域的研究热点。转录因子等调控因子通过识别特定的DNA或RNA序列基序,在转录或转录后水平调控基因的激活与抑制,因此基序的从头识别(de novo motif finding)是理解转录调控机制的关键第一步。现有研究表明,调控基序通常长度较短(真核生物中约6-15 bp)且具有高度简并性,难以与背景序列区分;传统基序识别方法多基于位置特异性评分矩阵(PSSM),通过Gibbs采样或期望最大化等迭代算法优化结合位点模型,但这类方法存在计算效率低、依赖初始候选序列选择、难以处理大规模数据集(如ChIP-seq实验产生的数千个非编码区域)的局限性,且在处理哺乳动物等复杂基因组的调控数据时扩展性不足,无法充分利用全基因组的定量调控证据。
针对上述领域内未解决的核心问题,即现有基序识别方法无法高效整合全基因组定量证据、处理大规模调控数据集的能力有限,本文提出了一种名为cERMIT的计算工具,基于全基因组的定量调控证据进行基序识别,无需预筛选候选序列,可高效适配ChIP-chip、ChIP-seq等大规模实验数据,为转录调控基序的识别提供了新的技术框架,具有重要的学术价值与应用前景。
2. 文献综述解析
作者对领域内现有基序识别研究的分类维度为核心算法策略,主要分为三类:一是基于位置特异性评分矩阵迭代优化的经典方法,二是基于简并共识序列的过表达分析方法,三是整合高通量实验数据的定量分析方法。
基于位置特异性评分矩阵迭代优化的方法(如AlignACE、MEME)通过迭代更新结合位点的亲和力模型,能较准确地描述转录因子结合位点的特征,这类方法在酵母等简单基因组的小规模数据集中表现出一定有效性,但存在计算复杂度高、易陷入局部最优、难以处理大规模数据集的局限性,且需预筛选候选序列,导致对低丰度、高简并性基序的识别能力不足。基于简并共识序列的方法(如Weeder)通过穷尽搜索与背景序列相比过表达的寡聚体,无需显式生成序列模型,但对背景序列的区分能力较弱,且在处理复杂基因组时计算效率低下。整合高通量实验数据的定量方法(如REDUCE、MatrixReduce、DRIM、Amadeus)则利用ChIP-chip、表达谱等定量数据辅助基序识别,其中REDUCE和MatrixReduce直接整合全基因组数据,无需预定义阈值,但DRIM和Amadeus仍需推断正负样本的cutoff,且这类方法在处理哺乳动物ChIP-seq等超大规模数据集时的扩展性仍有待提升。
通过对比现有研究的局限性,本文的创新价值凸显:cERMIT无需预筛选候选序列或推断阈值,直接利用所有序列区域的定量调控证据进行基序搜索,通过后缀数组数据结构实现高效的序列匹配,计算效率支持处理包含数千个非编码区域的哺乳动物ChIP-seq实验;同时在 curated ChIP-chip金标准数据集上的性能显著优于现有方法,填补了大规模调控数据集基序高效、准确识别的技术空白,为转录调控研究提供了更强大的计算工具。
3. 研究思路总结与详细解析
本文的研究目标是开发一种高效、可扩展的全基因组基序识别工具,核心科学问题是如何利用全基因组的定量调控证据,无需预筛选候选序列即可准确识别转录调控基序,技术路线遵循“算法框架构建→金标准数据集验证→大规模测序数据集扩展→跨类型数据通用性验证”的闭环逻辑,确保方法的准确性、扩展性与通用性。
3.1 cERMIT算法框架构建
实验目的:构建基于全基因组定量调控证据的基序识别算法框架,实现高效搜索与可扩展性。
方法细节:以推定调控区域集合S和对应的调控证据得分E为输入,采用借鉴基因集富集分析的目标函数,计算每个候选基序对应的正调控区域的聚合证据得分,该得分通过中心化和缩放的平均值与随机组比较,反映基序与调控证据的关联程度;从所有可能的5-mer序列作为种子,通过贪婪搜索策略迭代优化基序的长度(扩展、截断)和简并性(单碱基简并度调整);利用后缀数组数据结构实现快速的序列匹配,确保在大规模序列集中高效检测基序的所有出现位置。
结果解读:算法可输出灵活长度的简并共识序列基序,并可通过后处理步骤将高得分候选转换为位置特异性评分矩阵;目标函数无需预定义阈值,能有效整合全基因组的调控证据,计算效率显著提升,在标准单处理器工作站上,酵母转录因子的典型分析约1分钟/次,人类ChIP-seq的约35000个区域分析约2-5分钟/次。

产品关联:文献未提及具体实验产品,领域常规使用Python、R等计算工具及MAQ、F-seq等测序数据处理软件。
3.2 酵母ChIP-chip金标准数据集性能验证
实验目的:在已有的金标准数据集上验证cERMIT的性能优于现有基序识别方法。
方法细节:使用Young实验室的352个酵母ChIP-chip实验数据集,涵盖203个转录因子(82个转录因子包含多条件数据),以已知的文献共识基序为参考,采用Harbison等提出的位置特异性评分矩阵相似性度量(阈值设为0.75)评估预测性能;分别在三个子集上与现有方法对比:156个含已知共识的实验(80个转录因子)、150个与Amadeus研究重叠的实验(77个转录因子)、44个与DRIM研究重叠的实验(36个转录因子)。
结果解读:在156个实验子集上,cERMIT成功识别的基序数量显著高于AlignACE、MEME、MEME-c、Converge、MD-scan、PRIORITY等方法,即使不利用物种间保守性的ERMIT版本,性能也优于依赖保守性的方法;在150个实验子集上,允许前4个预测结果时,cERMIT成功识别100个条件(58个转录因子),显著高于Amadeus的78个条件(53个转录因子);在44个实验子集上,cERMIT前2个预测的成功率为83%(n=36,P<0.01),显著高于DRIM的53%(n=36,P<0.01)。
产品关联:文献未提及具体实验产品,领域常规使用ChIP-chip实验平台及相关生物信息学分析软件。
3.3 哺乳动物ChIP-seq大规模数据集应用验证
实验目的:验证cERMIT在哺乳动物大规模ChIP-seq数据集上的可扩展性与准确性。
方法细节:处理6个人类转录因子ChIP-seq数据集(STAT1、CTCF、SRF、GABP、FoxA1、NRSF)和12个小鼠胚胎干细胞ChIP-seq数据集(cMyc、nMyc、E2f1等);采用两种方式定义推定调控区域:一是基于DNaseI超敏感位点(DHS)的开放染色质区域,二是基于多因子ChIP-seq峰的“ensemble”集合;通过与TRANSFAC数据库中的已知位置特异性评分矩阵比较评估预测准确性。
结果解读:在人类数据集上,两种调控区域定义方式下cERMIT均能准确识别已知基序,其中CTCF基序通过前25000个DHS峰成功识别;在小鼠数据集上,cERMIT对所有因子均能恢复与已知基序高度相似的预测,对于无已知共识的Zfx,预测结果与Weeder、NestedMICA等方法一致;对于噪声较大的E2F数据集,cERMIT识别出类似部分共识的GC富集基序,且在Sox2数据集中能区分Sox2与Oct4的共结合基序,体现出处理复杂调控信号的优势。
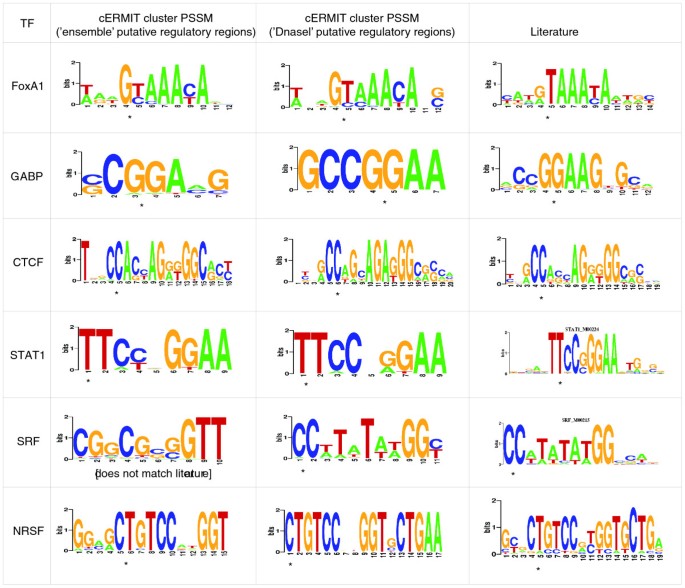
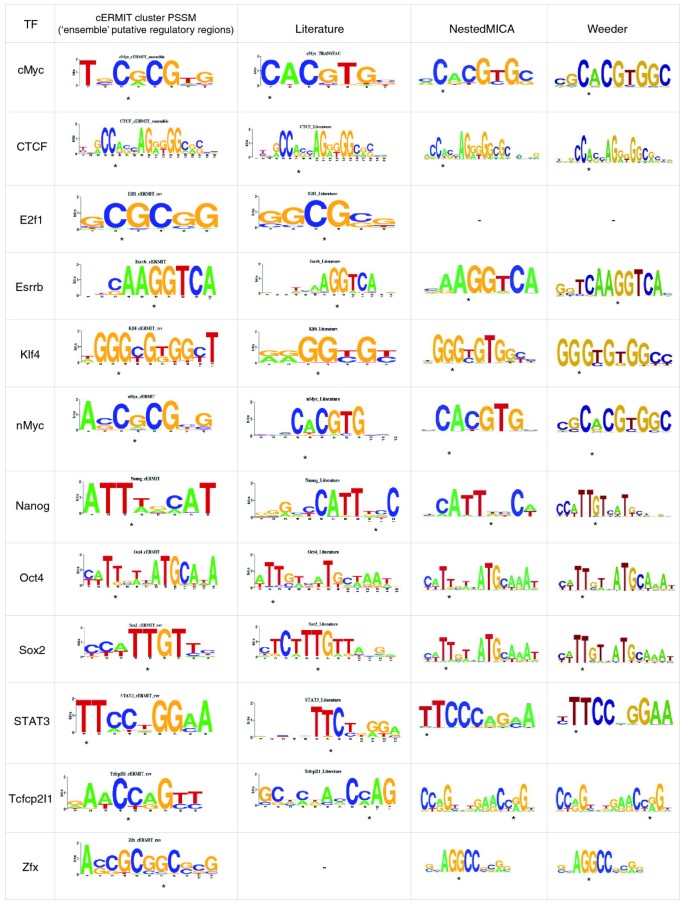
产品关联:文献未提及具体实验产品,领域常规使用Illumina等高通量测序平台及MAQ、F-seq等测序数据处理软件。
3.4 microRNA过表达实验通用性验证
实验目的:验证cERMIT在间接调控数据(如miRNA过表达的表达谱数据)中的通用性。
方法细节:使用5个microRNA(hsa-let-7b、hsa-miR-1、hsa-miR-155、hsa-miR-16、hsa-miR-30a)过表达的mRNA和蛋白质表达数据集,以基因3"UTR区域为调控区域,表达变化的log倍数为调控证据得分,比较cERMIT预测基序与经典miRNA种子序列(2-7、1-7、2-8、1-8位互补序列)的得分。
结果解读:cERMIT预测的基序得分均高于经典种子序列,除hsa-let-7b外,预测基序为种子序列的轻微变体;在蛋白质组数据中成功识别所有5个miRNA的结合基序,表明其可有效处理间接调控的表达数据,具有广泛的应用场景。
产品关联:文献未提及具体实验产品,领域常规使用microarray、蛋白质质谱平台及相关数据分析软件。
4. Biomarker研究及发现成果解析
本文涉及的Biomarker为功能性转录调控基序,包括转录因子结合基序和miRNA结合基序,属于调控元件类Biomarker,其筛选与验证逻辑为:基于全基因组定量调控证据(ChIP-chip P值、ChIP-seq读段计数、表达变化log倍数),通过cERMIT算法的贪婪搜索与目标函数评分筛选高得分基序,再通过与已知文献共识基序的位置特异性评分矩阵相似性比较、跨数据集一致性验证进行验证,形成“全基因组证据筛选→算法优化→多维度验证”的完整逻辑链条。
基序的来源包括酵母基因上游调控区域、人类和小鼠的开放染色质区域或ChIP-seq峰区域、人类基因3"UTR区域;验证方法采用Harbison位置特异性评分矩阵相似性度量(阈值≥0.75)、与TRANSFAC/JASPAR数据库中的已知基序比对;特异性与敏感性方面,在酵母ChIP-chip数据集上,当采用P值 cutoff 10^-4时,真阳性率为76%(n=88,P<10^-4),假阳性率为26%(n=68,P<10^-4),且能识别约25%其他方法无法检测的基序;在人类ChIP-seq数据集中,对STAT1、CTCF等因子的基序识别与已知共识的相似性均≥0.75,特异性与敏感性兼具。
核心成果提炼:首次提出基于全基因组定量调控证据的无阈值基序识别框架cERMIT,实现了对大规模ChIP-seq数据集的高效处理,计算效率与识别准确性显著优于现有方法;在酵母ChIP-chip金标准数据集上成功识别多个其他方法无法检测的低丰度、高简并性基序,真阳性率达76%;验证了该方法在转录因子结合基序、miRNA结合基序识别中的通用性,为复杂基因组的转录调控解析提供了高效工具;创新性在于无需预筛选候选序列,直接整合全基因组调控证据,打破了传统方法在处理大规模数据时的瓶颈,为后续调控基序的功能验证与疾病相关调控机制研究提供了重要基础。